PANEL DE DISCUSION |
L.Carreras. Servicio de Nefrología.
Hospital Princeps d'Espanya.
CSUB. Barcelona. Espańa
e-mail: lcarreras@mail.cinet.es
Síndrome Hemolítico Urémico (SHU) y Púrpura Trombótica Trombocitopénica (PTT) se consideran como formas agudas de microangiopatía trombótica (MAT) que pueden afectar, respectivamente y de forma predominante al rińón, en el primer caso, o al Sistema Nervioso Central en el segundo. Su común patogenia y respuesta similar al tratamiento parecen confirmarlo. Nuestra experiencia, como Servicio de Nefrología de adultos, queda limitada al SHU. Desde 1974 hasta enero de 2000, en un hospital de referencia para una población de alrededor de 1.000.000 personas, hemos diagnosticado de SHU un total de 41 pacientes de los que 18 eran mujeres y 23 varones, con edades comprendidas entre los 18 y los 69 ańos, con un promedio de 35,05 ± 13.51 ańos.
Posteriormente, el estudio de factores genéticos predisponentes nos ha llevado a estudiar a otros cinco pacientes, dos mujeres y tres varones que, con edades comprendidas entre los 2 y los 52 ańos, fueron tratados inicialmente en otros hospitales por SHU.
La aparición súbita fue la norma, excepto en una ocasión cuyo inicio fue insidioso, precediendo la alteración renal, proteinuria/ hematuria en dos meses, a la aparición de trombocitopenia y anemia hemolítica. Los transtornos de coagulación dieron lugar al primer síntoma de los pacientes: Hematuria macroscópica en 5, epistaxis en 2, gingivorragia en 1, equimosis en1, hemoptisis en 1 y metrorragias, que requirieron histerectomía en otro.
En ocho casos el paciente había consultado, inicialmente, por astenia y en otros cinco sólo cabía seńalar lo que fue interpretado como proceso catarral agudo. En una ocasión fue la instauración brusca de anuria, tan absoluta que remedaba un proceso obstructivo y que la ulterior biopsia diagnosticó como total necrosis cortical.
Han sido numerosos los mecanismos patogenéticos que se han invocado en la aparición de MAT: (Fig. 1),
| Dańo endotelial Alteraciones agregación plaquetar Factores genéticos |
En el momento de enumerar los posibles factores etiológicos (Fig. 2), la presencia exclusiva de adultos en nuestra serie discrepa de su asociación a cuadros diarréicos por E. Coli (Shiga toxin)9.
| HTA maligna: | 8 |
| Esclerodermia: | 1 |
| Anticonceptivos: | 8 |
| Postparto: | 3 |
| Forma familiar: | 6 |
| HIV: | 2 |
| Neoplasia de pulmón: | 1 |
| Ciclosporina: | 1 |
| Anticuerpos antiKidd: | 1 |
| E. Coli: | 1 |
Incluso la mayor incidencia estacional, estival, hallada en nińos, no aparece
en ella. Unicamente seis pacientes iniciaron su SHU en verano y, en dos, que
presentaban antecedentes inmediatos de proceso diarréico, sus coprocultivos
resultaron negativos. El E.Coli apareció en una sola ocasión, en una recidiva
post trasplante renal. Hipertensión arterial (HTA) maligna se constató en nueve
pacientes, uno de los cuales fue diagnosticado de esclerodermia. Ocho de las enfermas se hallaba bajo
anticonceptivos orales10 en el momento de desarrollar su SHU, tres
de las cuales presentaban HTA maligna. Otras tres pacientes lo presentaron
inmediatamente después del parto11. Dos de los pacientes eran HIV
positivos12, uno de los cuales se encontraba bajo tratamiento
antirretroviral. La aparición de SHU inmediatamente después de una hemóliis
post transfusional en paciente con anticuerpos antiKidd, madre de dos hijos
Kidd positivos, induce a suponer cierta relación con su hemólisis. La
ciclosporina13 no sólo ocasionó la aparición de novo en un paciente
trasplantado renal, sino que fue, sin
duda, responsable en un paciente que, después de un trasplante de médula ósea,
desarrolló una enfermedad injerto contra huésped donde fue utilizada. Su
inmediata suspensión permitió la remisión del proceso. Otros dos episodios
aparecieron, de novo, en pacientes trasplantados bajo tacrolimus14.
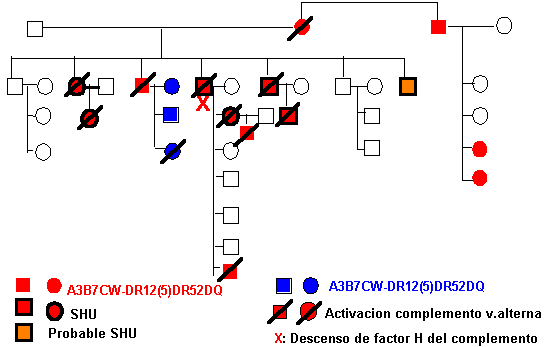
El SHU ha sido de gran incidencia en una familia de
la que hemos estudiado 24 miembros a lo largo de cuatro generaciones más 7
familiares no emparentados5 (Fig. 3). Se demostró cómo en todos los
casos de SHU, los pacientes, seis en total, compartían el mismo haplotipo HLA y
presentaban un descenso del complemento por activación de la vía alterna persistentemente*.
Otros cuatro familiares, con edades comprendidas entre los 7 meses y los 86 ańos,
portadores del mismo haplotipo, también eran hipocomplementémicos, si bien no han presentado, por el momento, signos de microangiopatía trombótica.
El HUS afectaba a ambos sexos y en edades entre 4 meses y 52 ańos. En dos casos se puede invocar una causa desencadenante, la toma de anticonceptivos orales en una paciente de 32 ańos y la presencia de una adenocarcinoma pulmonar15 en un hermano, varón de 35 ańos. Debe seńalarse la existencia de un tercer caso, consumidor habitual de bebidas con quinina, lo que ha sido invocado como relacionado con la aparición de MAT16.
La presencia de un haplotipo común (A3B7CW-DR12DR52DQ) en todos los pacientes llevó a sospechar que este tipo de herencia, autosómica dominante, podría estar relacionado con alguna alteración genética en las proximidades del complejo de histocompatibilidad mayor, en el 6ş cromosoma, pero el simulation linkage analyisis no resultó significativo17. La presencia en ellos de hipocomplementemia por activación de la vía alterna motivó el estudio del factor H (inmunodifusión radial) en estos pacientes que, con excepción de uno sólo, resultó siempre normal. Dicho haplotipo no se ha repetido en ninguno de los pacientes restantes afectos de HUS, ni guarda relación con lo publicado18,19. Pero sí ha tenido cierta incidencia el A29 y B35, presentes en diez de los 30 pacientes estudiados. En cuatro coincidían ambos.
La afectación familiar se daba también en dos pacientes, mujeres, una de las cuales, hipocomplementémica, había desarrollado el proceso cuando se hallaba bajo anticonceptivos orales, y ambas tenían primas diagnosticadas en otros hospitales de HUS. Incluso se investigó en dos hermanos gemelos univitelinos que presentaron este proceso en episodios separados por más de dos ańos. En ningún caso pudo hallarse conexión entre ellos.
En un total de trece pacientes se observó descenso del complemento20 por activación de la vía alterna. La necrosis tisular que sufrían, así como los inevitables problemas de biocompatibilidad con las membranas de diálisis podrían ser la causa de su hipocomplementemia. El factor H, estudiado en los pacientes de la afectación familiar ya mencionados y en otro paciente de 24 ańos ajeno a ella, fue normal en todos los casos excepto en uno.
La relación entre HUS y PTT quedó probada en 17 pacientes que tuvieron alguna manifestación extrarrenal (Fig. 4).
| Miocardiopatía | 4 |
| Convulsiones: | 4 |
| Parálisis facial: | 1 |
| Foco irritativo (EEG): | 1 |
| Elevación amilasa pancreática: | 4 |
| Púrpura: | 2 |
| IAM + AVC: | 1 |
La afectación cerebral24 ocasionó cuadros convulsivos en cuatro pacientes que carecían de antecedentes de previos problemas comiciales que necesitaron tratamiento, obteniéndose remisión total. Un paciente presentó parálisis facial y otro tuvo focalidad irritativa en el EEG.
Se observó dolor abdominal con elevación de amilasa pancreática en cuatro pacientes, pero su aumento en la insuficiencia renal y la concomitancia de tratamiento con corticoides y diuréticos impidió atribuir el cuadro a una afectación pancreática Lesiones maleolares purpúricas aparecieron en dos pacientes durante la fase aguda de su HUS. Biopsiadas fueron etiquetadas como de púrpura leucocitoclástica.
Cabe seńalar que un paciente sufrió infarto agudo de miocardio y accidente vasculocerebral que le ocasionaron la muerte, al día siguiente de haberse diagnosticado su HUS
La hemodiálisis fue necesaria en todos excepto nueve pacientes. Uno falleció antes de poder iniciar el tratamiento. Se utilizó plasma fresco y/o plasmaféresis en 24 pacientes. De ellos 14 recibieron ambos tratamientos, 6 sólo plasmaféresis y cuatro sólo plasma fresco. El tratamiento se prolongó hasta alcanzar la curación o mientras haptoglobinas y LDH seńalaban la presencia de hemólisis. En 14 se ańadió misoprostol. Sólo tres pacientes recuperaron una función renal normal.
La adicción de vitamina E masiva en 3 pacientes no ofreció mejoría remarcable.
En una ocasión se utilizó vincristina donde ya habían fracasado plasmaféresis y plasma fresco sin que tampoco ofreciese resultados positivos.
Se practicó trasplante renal en ocho pacientes, recidivando el SHU en seis de ellos, uno de los cuales estaba bajo ciclosporina. Mientras que apareció de novo en tres: Uno bajo ciclosporina y dos en tratamiento con tacrolimus
La severidad del proceso es evidente.
Seis pacientes fallecieron durante su posterior estancia en hemodiálisis periódica y uno paciente falleció en la fase aguda por infarto agudo de miocardio.
Ocho alcanzaron, y aún mantienen entre 1 a 24 ańos después, remisión completa. Veintidós alcanzaron la fase final de su insuficiencia renal quedando en programa de hemodiálisis periódica hasta su posterior trasplante renal. El resto se halla en diferentes fases de insuficiencia renal.
Discusión.
La relación entre SHU y PTT se reafirma ante el hallazgo de manifestaciones extrarrenales en 17 pacientes. Se trata de un problema multifactorial en el que nuestra experiencia nos permite incidir sobre los aspectos genéticos. Parece obvia la predisposición genética de la amplia familia reseńada. Seis casos confirmados, ampliamente separados en el tiempo lo que descarta un problema epidémico, y un séptimo posible lo ratifican. Sin embargo, no se ha podido probar su relación con el haplotipo involucrado ni con el descenso del complemento hallado en sus miembros. La normalidad del factor H en casi todos ellos discrepa de lo expuesto hasta ahora. Tampoco el estudio de los otros tres grupos familiares con afectación de dos miembros en cada uno ha aportado datos esclarecedores. Es necesario estudiar el papel de otros moduladores del complemento, como el factor I y de las alteraciones en la degradación del factor de von Willebrand que parecen más evidentes en la PTT.
En un intento de hallar factores pronósticos el examen anatomopatológico25 nos ha permitido relacionar la severidad de la insuficiencia renal con la isquemia glomerular y con la MAT yuxtaglomerular. Pero no existía una aparente relación entre deterioro funcional y MAT glomerular Eran escasas las lesiones en arterias interlobulillares
La evolución, si bien su gravedad es manifiesta26, 27, ha sido variable. De los ocho pacientes que recuperaron función renal, tres habían presentado un SHU posparto, que tradicionalmente se asocia con mejor pronóstico, y su satisfactoria evolución se alcanzó exclusivamente con tratamiento conservador, hemodiálisis. Si bien su bajo número impide precisar ningún dato, si cabe seńalar que, en conjunto eran pacientes jóvenes, 18- 34 ańos, excepto uno de 66 y su creatinina inicial era relativamente menor que la del resto, entre 140 y 529 mmol/l, de modo que cinco de ellos no requirieron hemodiálisis. Contrasta todo ello con las elevadas cifras de creatinina (118- 1673 mmol/l, X= 661 ± 437mmol/l) o los requerimientos de diálisis que inicialmente presentaban los pacientes que no obtuvieron remisión del proceso.
Los resultados de los diversos tratamientos ensayados no han sido satisfactorios. Aunque la plasmaféresis28 se ha mostrado más eficaz que la exclusiva administración de plasma fresco en su acción sobre la hemólisis, no ha impedido la aparición de recidivas ni la progresión hacia la insuficiencia renal. La administración de prednisona en once pacientes, misoprostol en 14 y dosis masivas de vitamina E en tres o vincristina en uno, no permite establecer ninguna relación con la evolución del proceso.
El trasplante renal sigue siendo una opción de alto riesgo por sus frecuentes recidivas. La introducción de micofenolato mofetil parece, por el momento, la más adecuada de las opciones.
Bibliografía:
1. Furlan M, Robles R, Galbusera M et al. Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the haemolytic-uremic syndrome. N Engl J Med 1998; 339:1578
2. Tsai H-M, Lian EC-Y. Antibodies to von Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998; 339: 1585
3. Ballermann BJ, Endothelial cell activation. Kidney Int 1998; 53: 1810
4. Mitra D, Jaffe EA, Weksler B et al. Thrombotic thrombocytopenic purpura and sporadic haemolytic uremic syndrome plasmas induce apoptosis in restricted lineages of human microvascular endothelial cells. Blood 1997; 89: 1224
5. Carreras L, Romero R, Requesens C et al. Familial hypocomplementemic hemolytic uremic syndrome with HLA A3B7 haplotype JAMA 1981; 245: 602
6. Rougier N, Kazatchkine MD, Rougier JP et al. Human complement factor H deficiency associated with haemolytic uremic syndrome. J Am Soc Nephrol 1998; 9: 18- 26
7. Warwicker P, Goodship THJ, Donne RL et al. Genetic studies into inherited and sporadic haemolytic uremic syndrome. Kidney Int 1998; 53: 836
8. Pichette V. Querin S, Schurch W et al. Familial haemolytic-uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis 1994; 24: 936
9. Kaplan BS, Meyers KE, Schulman SL. The pathogenesis and treatment of haemolytic uremic syndrome. J Am Soc Nephrol 1998; 9: 1126
10. Hauglustaine D, van Damme B, van Renterghem Y, Michielsen P. Recurrent haemolytic-uremic syndrome during oral contraception. Clin Nephrol 1981; 15: 148
11. Weiner CP. Thrombotic microangiopathy in pregnancy and the postpartum period. Semin Hematol 1987; 24: 119
12. Thompson CE, Damon LE, Ries CA, Linker CA. Thrombotic microangiopathies in the 1980s. Clinical features, response to treatment , and the impact of the human immunodeficiency virus epidemic. Blood 1992; 80: 1980
13. Myers BD. Cyclosporine nephrotoxicity. Kidney Int 1986; 30: 964
14. Trimarchi HM, Truong LD, Brennan S et al. FK506-associated thrombotic microangiopathy. Transplantation 1999; 67: 539
15. Podzamger D, Carreras L, Condom E, Baucells JM, Vidaller A. Microangiopathic hemolytic anemia associated with pulmonary adenocarcinoma JAMA 1985: 254:18, 2554
16. Gottschall JL, Neahring B, McFarland JG et al. Quinine-induced immune thrombocytopenia with haemolytic uremic syndrome: Clinical and serological findings in nine patients and review of literature. Am J Hematol 1994; 47: 283
17. Carreras L, Volpini V, Poveda R et al. Familial Hemolytic Uremic Syndrome. Simulation linkage analysis. En Hereditary Kidney Disease. A.Sessa. Contributions to Nephrology. Karger Ed. Basel, 1997. 196
18. Joseph G, Smith KJ, Hadley TJ et al. HLA-DR53 protects against thrombotic thrombocytopenic purpura/adult haemolytic uremic syndrome. Am J Hematol 1994; 41: 189
19. Sheth KJ, Gill JC, Leichter HE et al. Increased incidence of HLA-B40 group antigens in children with haemolytic-uremic syndrome. Nephron 1994; 68: 433
20. Noris M, Ruggenenti P, Perna A et al. Hypocomplementemia discloses genetic predisposition to haemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Role of factor H abnormalities. J Am Soc Nephrol 1999; 10: 281
21. Abu Arafeh I, Gray E, Youngson G et al. Myocarditis and haemolytic uremic síndrome. Arch Dis Child 1995; 72: 46 22. Walker AM, Benson LN, Wilson GJ, Arbus GS. Cardiomyopatia: a late complication of haemolytic uremic syndrome. Pediatr Nephrol 1997; 11: 221
23. Fuertes J, Torre A, Merino JL et al. Miocardiopatía dilatada y síndrome hemolítico urémico. Nefrología 1998; 6: 498
24. Siegler RL. Spectrum of extrarrenal involvement in postdiarrheal haemolytic uremic syndrome. J Pediatr 1994; 125: 511
25. Robres AF, Carreras L, Poveda R et al. SHU/PTT: Caracteristiques cliniques i evolutives.. XII Reunió Anual de la Soc. Catalana de Nefrología. La Seu d’Urgell, 1996
26. Conlon PJ, Howell DN, Macick G et al. The renal manifestations and outcome of thrombotic thrombocytopenic purpura/haemolytic uremic syndrome in adults. Nephrol Dial Transplant 1995; 10: 1189
27. Matsumae T, Takebayashi S, Naito S. The clinico- pathological characteristics and outcome in haemolytic uremic syndrome of adults. Clin Nephrol 1996; 45: 153
28. Hollenbeck M, Kutkuhn B, Aul C et al. Haemolytic-uremic syndrome and thrombotic-thrombocytopenic purpura in adults: clinical findings and prognostic factors for death and end-stage renal disease. Nephrol Dial Transplant 1998; 13: 76