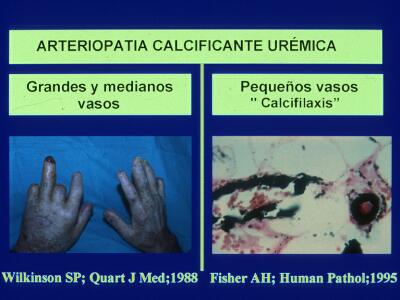
Figura 1. Arteriopatķa calcificante urémica. Izda: Amputación provocada por isquemia distal. Dcha: Depósito de calcio (azul oscuro) en vasos de la piel en una paciente con calcifilaxis.
PANEL DE DISCUSION |
Elvira Fernįndez Girįldez
Servicio de Nefrologķa del Hospital Universitario Arnau de Vilanova de Lleida y Departamento de Medicina de la Facultad de Medicina de la Universidad de Lleida. Spain
Introducción
En las śltimas décadas del siglo XX, uno de los retos mįs estimulantes para el nefrólogo ha sido el control del hiperparatiroidismo secundario (HPs). Hoy sabemos que el precio a pagar esta siendo un incremento de las calcificaciones de tejidos blandos de las cuales las que tienen mayor repercusión clķnica son las calcificaciones vasculares. (Fig.1, Fig.2). Ademįs, el uso generalizado de derivados de la vitamina D y compuestos de calcio han contribuido a inclinar la balanza del alto al bajo turnover óseo , observįndose un incremento en la prevalencia de enfermedad ósea adinįmica (EOA) (1).
Figura 1. Arteriopatķa calcificante urémica. Izda: Amputación provocada por isquemia distal. Dcha: Depósito de calcio (azul oscuro) en vasos de la piel en una paciente con calcifilaxis.
Figura 2. Infarto intestinal masivo en un paciente en diįlisis con calcificaciones vasculares mesentéricas.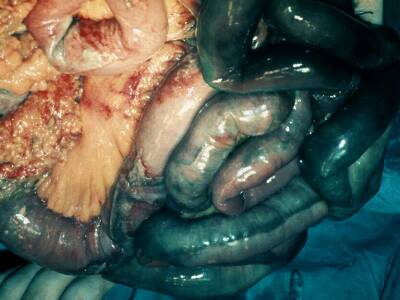
El incremento en la patologķa vascular y la mayor prevalencia de EOA se debe también el cambio en las caracterķsticas de la población con un mayor porcentaje de pacientes diabéticos y de edad avanzada. Todo ello nos obliga a replantearnos el tratamiento del disbalance fosfo-cįlcico del paciente con insuficiencia renal crónica (IRC) mirando no sólo hacia el hueso sino también hacia los vasos y el corazón.
La importancia del control del metabolismo fosfo-cįlcico en los pacientes con IRC radica en dos hechos probados:
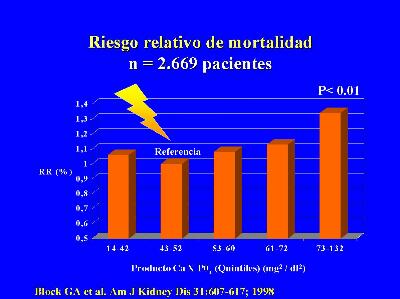
Figura 3. Riesgo relativo de mortalidad segśn diferentes rangos de producto
Ca X PO4 (quintiles). Adaptada de Block GA et al. (Ref. 2).
y 2. Su corrección induce mejorķa clķnica (3,4) (Fig.4, Fig.5).
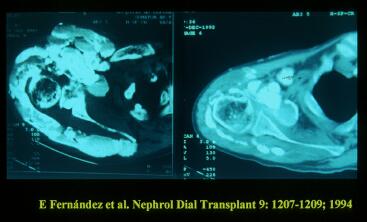
Figura 4. Calcinosis tumoral en la articulación del hombro en un paciente en diįlisis. Antes (Izda) y después (Dcha) de provocar balance negativo de calcio y fósforo mediante diįlisis diaria con dializado bajo en calcio. Puede observarse la casi completa resolución del depósito masivo de calcio.
Figura 5. Izda: Paciente trasplantada con calcifilaxis en ambas piernas. Dcha: Curación de las ślceras isquémicas después de la paratiroidectomia.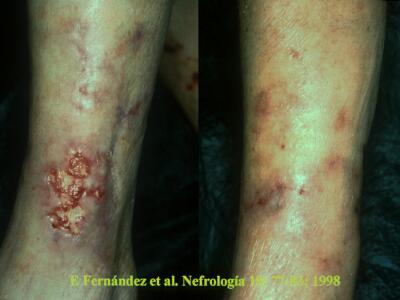
A. ENFERMEDAD ÓSEA DE ALTO TURNOVER: Hiperparatiroidismo secundario
A.M. Parfitt en su interesante articulo de revisión "The Hyperparathyroidism of chronic renal failure: a disorder of growth" (5) dice: "The rate of hormone secretion by the parathyroid, as of any endocrine gland, is the product of the average of hormone secretion by each individual cell and the total number of participating cells, but much more attention has been given to the regulation of hormone secretion than to the regulation of cell number . Ademįs, todas las medidas terapéuticas dirigidas a frenar la secreción de la parathormona implican una sobrecarga de calcio y fósforo imposible de eliminar mediante diįlisis o en pacientes con IRC avanzada (6).
Este anįlisis realista plantea la siguiente cuestión:
æEs posible tratar el HPs sin provocar un balance positivo de calcio y fósforo?
El objetivo del tratamiento del dismetabolismo fosfo-cįlcico en los pacientes con IRC debe ser conseguir un balance similar al de un sujeto sano de su misma edad. Para el fósforo este es neutro o ligeramente negativo en edades comprendidas entre 20 y 53 ańos y el balance de calcio tiende a ser negativo por encima de los 35 ańos .
La pérdida de nefronas funcionantes altera muy precozmente este equilibrio. El balance de fósforo es positivo desde etapas muy iniciales de insuficiencia renal y se incrementa cuando se inicia el tratamiento con diįlisis. Tanto la hemodiįlisis como la diįlisis peritoneal son insuficientes para eliminar el exceso de fósforo ingerido, si el paciente recibe una dieta adecuada. Ademįs, el tratamiento con calcitriol aumenta de un 60% a un 86% la absorción de fosfato.
La absorción intestinal de calcio esta disminuida en la IRC por el déficit de vitamina D pero el balance de calcio puede ser positivo, incluso antes del inicio de la diįlisis si el paciente ingiere una dieta rica en calcio o se administran compuestos de calcio y vitamina D. Tanto la hemodiįlisis como la CAPD provocan un balance positivo de calcio si se utilizan dializados con concentración de calcio igual o superior a 2.5 mEq/l (6).
Para analizar en profundidad el balance de calcio y fósforo en la IRC remito al lector a la excelente revisión de Chen H. Hsu (6) o a la lectura de su conferencia en el presente Congreso :
Dilemas del presente para normalizar el balance de fósforo
proteica conlleva riesgo de desnutrición. Por otro lado son pocos los pacientes que se ajustan a las recomendaciones dietéticas (7,8).
Ø La administración de quelantes del fósforo tiene efectos adversos: Los compuestos de aluminio han demostrado sobradamente tener un efecto tóxico sobre el hueso. Los compuestos de calcio favorecen la hipercalcemia y su uso se ha relacionado con un riesgo mayor de calcificación de tejidos blandos (9,10).
Ø El incremento en la eficacia de la eliminación mediante diįlisis es limitada: Una vez que el fósforo plasmįtico ha alcanzado el nadir, ni las membranas de alta permeabilidad ni el aumento de la duración de la sesión de diįlisis mejoran la eficacia de su eliminación (11,12).
Dilemas del presente para normalizar el balance de calcio
Ø La disminución de la concentración de calcio del dializado estimula la función paratiroidea (13,14) (Fig.6).
Ø La modificación en el manejo del tratamiento con derivados de la vitamina D (administración en bolus) tiene un ķndice muy elevado de fallos provocados por la severidad del hiperparatiroidismo y el desarrollo de hipercalcemia e hiperfosforemia (15): La demostración de que el calcitriol interaccionando con su receptor (VDR), ejerce una acción inhibidora directa sobre el gen de la PTH, abrió una interesante vķa terapéutica. Esta se basó en el razonamiento de que niveles suprafisiológicos de calcitriol podrķan vencer la resistencia de la célula paratiroidea al calcitriol en la uremia. No se ha demostrado superioridad de ninguna de las vķas ni de las formas de administración. Ademįs, la refractariedad al tratamiento médico en glįndulas hiperplįsicas tiene una base molecular: el crecimiento autónomo de clonas celulares que presentan disminución de la densidad de los receptores de calcio extracelular (CaR) de VDR y probablemente también del receptor multifuncional endocķtico, la megalin/gp330, que parece tener funciones de sensor de calcio (16-19).
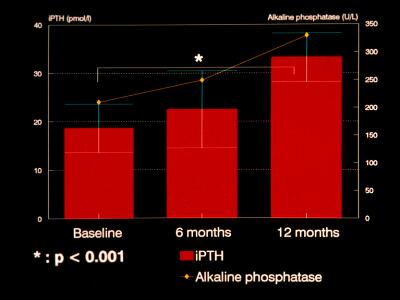
Figura 6. Incremento en los niveles de PTH y fosfatasa alcalina después de 6 y 12 meses de cambiar la concentración del dializado de 3.5 mEq/l a 2.5 mEq/l en una población de pacientes en hemodiįlisis.
En conclusión, con los esquemas terapéuticos empleados en las ultimas décadas del siglo XX, solo hemos conseguido "frenar" la función paratiroidea a expensas de un balance positivo de calcio y fósforo.
Estrategias de futuro en el tratamiento del HPs basadas en la evidencia cientķfica actual
1. Evitar el crecimiento glandular
2. Nuevos quelantes de fósforo
3. HD nocturna
4. Calciomiméticos
5. Anįlogos de la vitamina D
6. Nuevas técnicas para la reducción del tamańo glandular
7. Modulación de la función paratiroidea por terapia génica ?
1. Evitar el crecimiento glandular
Para poder evitar el crecimiento glandular es necesario conocer cual o cuales son los factores que lo ponen en marcha. Estį demostrado que el déficit de calcitriol, la hipocalcemia y la hiperfosforemia estimulan la secreción de parathormona y la proliferación celular (5,20,21). En varios estudios clķnicos las anomalķas detectadas mįs precozmente son el déficit de 1,25 (OH)2D3 y elevados niveles de PTH con niveles séricos normales de calcio y fósforo (22). Sin embargo, la normalidad de los niveles plasmįticos de calcio y fósforo no implica ausencia de alteración. A.Felsenfeld y M. Rodriguez (23) proponen que un fósforo sérico "normal" no debe ser considerado "apropiado" con niveles elevados de PTH.
Portale et al.(24) demostraron en individuos sanos y en etapas incipientes de IRC, que la manipulación del aporte de fósforo induce cambios reversibles en el calcitriol plasmįtico sin modificación en el fósforo sérico pero con un incremento en la fracción de excreción de fósforo (Fig.7). Existen resultados dispares en diversos estudios clķnicos modificando el aporte de fósforo de la dieta, probablemente por diferencias en la metodologķa (25-27). Para un detallado anįlisis del tema remito al lector a la conferencia de la Dra I. Martinez en este Congreso.
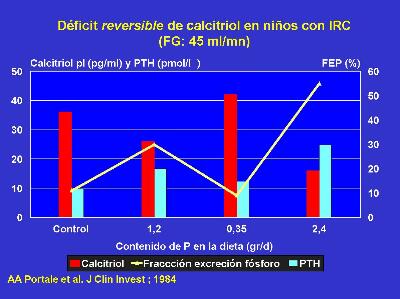
Figura 7. Efecto de la restricción (5 dķas) y suplemento de fósforo en la dieta, en nińos con IRC moderada, sobre la concentración plasmįtica de calcitriol, PTH y fracción de excreción de fósforo. Adaptado de Portale AA et al. (Ref. 24).
Existe una gran variabilidad en el grado de desarrollo del HPs, no explicada en su totalidad por los factores de riesgo conocidos, que sugiere influencia genética. Nosotros, en estudios previos, hemos demostrado la influencia del polimorfismo Bsm I del gen del VDR sobre los niveles de PTH y sobre el nivel plasmįtico de calcitriol en pacientes con IRC (28,29) (Fig.8).
Figura 8. Grįfico de "cajas" (programa informįtico SPSS) en el que se muestra los niveles plasmįticos de PTH en pacientes con diferentes rangos de función renal. El anįlisis estadķstico "Anova factorial general" tomando como variable dependiente el nivel de PTH, seleccionó las variables indicadas en los recuadros amarillos. Aclaramiento de creatinina = o > a 55 ml/mn (recuadro Izdo) y Aclaramiento de creatinina < A 55 ML/MN (RECUADRO DCHO). ADAPTADO DE MARCO MP ET AL. (REF. 29). CóMO PUEDE OBSERVARSE LOS CASOS CON HPS EXTREMO PERTENECEN MAYORITARIAMENTE AL GENOTIPO BB.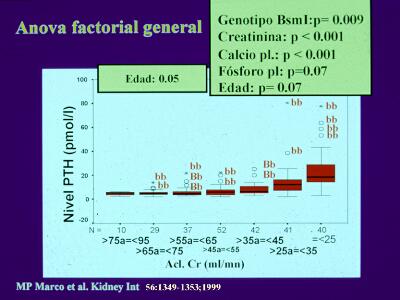
A medida que se deteriora la función renal existe un descenso de calcitriol irreversible y el nivel elevado de fósforo plasmįtico juega un papel central en el desarrollo del HPs (30,31). Por otro lado, estudios en animales de experimentación evidencian la resistencia del hueso a la acción de la PTH en la uremia (32,33) sin que se conozcan con esactitud los factores responsables de esta resistencia..
Por tanto, la estrategķa de futuro, teniendo en cuenta este mecanismo fisiopatológico, deberķa orientarse a evitar el crecimiento glandular:
2. Nuevos quelantes del fósforo
Los efectos secundarios de los quelantes clįsicos del fósforo, hipercalcemia con los compuestos de calcio y toxicidad por aluminio con los compuestos de aluminio esta justificando la bśsqueda de nuevos quelantes.
El nuevo compuesto, polyallylamine-hydrochloride (RenagelR) es un hidro-gel, resistente a la degradación digestiva y no absorbible. Ha creado enormes expectativas debido a que no contiene calcio ni aluminio. Un reciente estudio multicéntrico en 172 pacientes en hemodiįlisis demostró su efectividad bajando los niveles de fósforo plasmįtico con un descenso paralelo de los niveles de PTH. Otro adicional efecto beneficioso es el descenso en los niveles de colesterol y las LDL colesterol sin efecto en las HDL ni triglicéridos (35). Diversos estudios han confirmado su eficacia pero deberį probarse si son suficientemente efectivos como para ser utilizados como monoterįpia.
Otros productos como los compuestos de hierro o los inhibidores del "Na-P transporter" intestinal deben probar su eficacia y la ausencia de efectos indeseables.
3. HD nocturna
La hemodiįlisis lenta nocturna es una modalidad de diįlisis domiciliaria , conectada vķa módem con el hospital, desarrollada en el Wellesley Central Hospital de la Universidad de Toronto (Canada). Consiste en aumentar las frecuencias de las sesiones de diįlisis a 6-7 a la semana con una duración de 8 a 10 horas y parįmetros de diįlisis de baja eficacia. Esta técnica permite una mayor depuración de fósforo que la HD convencional. A largo plazo se logra un buen control del fósforo sin necesidad de captores de fósforo y con una dieta mįs liberal (36). Tiene el inconveniente de que requiere un catéter como acceso vascular para evitar accidentes y un sistema informįtico avanzado tanto en el hospital cómo en el domicilio del paciente. En el futuro deberan idearse fórmulas que permitan aumentar la frecuencia de diįlisis. Las limitaciones actuales son el acceso vascular, la interferencia con la actividad del paciente y el coste económico.
4. Calciomiméticos:
Recientemente, Brown y col. (37) han clonado un sensor del Ca++ extracelular (CaR) mayoritariamente expresado en las células paratiroideas, Células C productoras de calcitonina y tśbulo renal.
Responde a las variaciones de calcio plasmįtico induciendo sķntesis o inhibición de PTH.
Estos hechos han abierto una perspectiva terapéśtica de gran interés tanto en el hiperparatiroidismo primario como en el HPs. En la actualidad, se estįn desarrollando compuestos ("Calciomiméticos"), que activan el CaR en la región transmembrana, mientras que el Ca++ lo hace en el dominio extracelular. Por esta razón se les puede considerar "moduladores positivos" mas que "agonistas". Estos agentes han demostrado en IRC experimental que inhiben la proliferación paratiroidea y que suprimen la secreción de PTH mejorando la histologķa ósea (38,39). En pacientes con HPs se ha comprobado un efecto supresor agudo dosis-dependiente de la secreción de PTH sin efectos adversos, aunque se acompańó de un descenso del calcio iónico plasmįtico (40).
Es probable que el uso de estos agentes en el HPs requiera el uso de derivados de la vitamina D para evitar la hipocalcemia.
5. Anįlogos de la vitamina D
En la actualidad el objetivo de la investigación farmacéutica es conseguir derivados de la vitamina D con un efecto mįs selectivo sobre la célula paratiroidea que sobre la mucosa intestinal para evitar los episodios de hipercalcemia e hiperfosfatemia que limitan el uso del calcitriol. Fundamentalmente son tres los compuestos ensayados en el HPs:
Se requieren estudios en grupos amplios de pacientes y sobre todo a largo plazo para determinar las ventajas que apuntan los diferentes anįlogos. No debemos olvidar que las expectativas creadas sobre el menor efecto hipercalcemiante en estudios a corto plazo con la administración del calcitriol en bolus intravenosos , no se han cumplido cuando los pacientes se han tratado durante periodos mįs prolongados.
6. Nuevas técnicas para la reducción del tamańo glandular
En la actualidad la indicación de paratiroidectomķa se establece cuando ha fracasado el tratamiento médico. Como dice Parfitt (5), esta polķtica refleja el concepto erróneo de considerar como objetivo terapéutico el nivel de secreción glandular mas que el tamańo.
La técnica seguida sigue siendo objeto de controversia (50). Las estrategias que probablemente se desarrollaran en el futuro son:
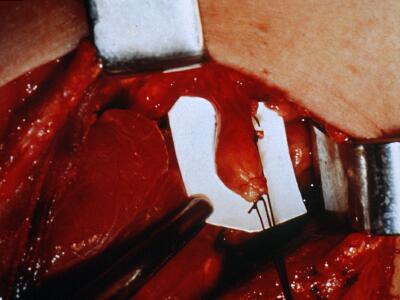
Figura 9. Técnica quirśrgica de paratiroidectomia en la que se muestra la disección de la paratiroides inferior Dcha respetando el pedķculo vascular. Posteriormente serį situada en el tejido subcutįneo.
En la actualidad se han conseguido transferir genes funcionales, utilizando cómo vector un adenovirus, en células paratiroideas humanas dispersas. También se ha logrado transferir el gen lacZ en glįndulas paratiroides de ratas por inyección directa del virus (16). Es probable que en el futuro sea técnicamente posible modular la función paratiroidea por terapia génica.
B. ENFERMEDAD ÓSEA DE BAJO TURNOVER
Enfermedad ósea adinįmica
En los śltimos ańos se esta observando un cambio en el espectro de los patrones de osteodistrofia, debido fundamentalmente a un incremento en la incidencia de EOA (entre el 40 y 71%) (52). Este incremento se ha atribuido a mśltiples factores (1):
Aunque quedan todavķa cuestiones por resolver en la enfermedad ósea de bajo turnover inducida por aluminio, probablemente en los próximos ańos asistiremos a una disminución de su prevalencia debido al impacto de los siguientes factores: 1.La aceptación de un nivel mįximo de aluminio de 2-3 mcg/l en el dializado; 2. La desaparición de los compuestos de aluminio como quelantes del fósforo; 3. El seguimiento de protocolos (DOQI) para tratar la ferropenia en pacientes tratados con Eritropoyetina.
Sin embargo, la EOA ha emergido en los śltimos ańos cómo una patologķa de la que se conoce su carįcter multifactorial pero se desconocen mśltiples aspectos relacionados con su expresividad clķnica y tratamiento. En los śltimos ańos se han planteado el siguiente dilema clķnico:
æLa EOA tiene consecuencias clķnicas y por tanto debe ser tratada o evitada?
Se le han atribuido 2 manifestaciones clķnicas relevantes :
· Mayor riesgo de fracturas: Existen varios estudios epidemiológicos que demuestran un riesgo elevado de fracturas en la población diįlisis (54). Sin embargo, no estį establecida una asociación clara con el tipo histológico de osteodistrņfia renal. AM Parfitt (55), en su excelente revisión sobre la estructura ósea en la enfermedad renal, concluye que el factor que mįs influye en el riesgo de fracturas, es el adelgazamiento generalizado de la cortical. Este factor, que ya estį presente antes del comienzo de diįlisis, serį el mayor determinante en el futuro del riesgo de fractura, independientemente del tipo histológico de osteodistrofia . Debido a que la PTH tiene un efecto anabólico sobre el hueso esponjoso y catabólico sobre el hueso cortical su recomendación es la prevención del HPs y recuerda el efecto beneficioso de la partiroidectomķa sobre la densidad ósea. Sin embargo, Stein et al. (56) encuentran que el riesgo de osteopenia es mayor en pacientes con trasplante previo, paratiroidectomizados y mujeres con amenorrea secundaria.
La división clįsica de la osteodistrófia renal en alto y bajo remodelado óseo, no contempla la coexistencia de osteoporosis. Esta enfermedad metabólica, relacionada fundamentalmente con la edad y con los déficits de hormonas sexuales tiene mayor riesgo de fracturas. Es razonable pensar, aunque no estį demostrado, que su prevalencia sea superior en la población de diįlisis caracterizada por una edad media elevada e hipogonadismo (57). Remito al lecor a la conferencia de la Dra T. Gonzalez en este Congreso.
· Mayor riesgo de hipercalcemia: Kurz et al. (58) demostraron la disminuida capacidad del hueso adinįmico para absorber la sobrecarga de calcio. En este mismo estudio, también se observa menor capacidad de absorción de calcio intestinal en mujeres en hemodiįlisis.
Dilemas del presente en el tratamiento de la enfermedad ósea adinįmica
Ø No disponibilidad de marcadores de remodelado óseo fiables y utilización de la PTH sérica cómo śnico indicador de respuesta al tratamiento. Un error clķnico frecuente es la insistencia en tratar el HPs sin tener en cuenta otros marcadores de remodelado óseo. Esto conduce a incrementar considerablemente el balance de calcio y fósforo en un paciente cuyo hueso no pude absorber ese exceso y consecuentemente favorece su depósito en tejidos blandos.
Ø El estķmulo de la función paratiroidea utilizando un dializado bajo en calcio y evitando los compuestos de calcio y derivados de la vitamina D (13,59), puede aumentar el riesgo de osteoporosis . Ademįs, los niveles moderadamente elevados de PTH aconsejados para mantener un turnover óseo adecuado en el grupo de mayor riesgo (pacientes ańosos ,diabéticos, tratados con diįlisis peritoneal ambulatoria continua) pueden tener consecuencias adversas cardiovasculares.
En conclusión, la EOA tiene consecuencias clķnicas y por tanto debe prevenirse y tratarse. En la actualidad no existe un tratamiento probadamente eficaz e inocuo.
Estrategias de futuro basadas en la evidencia cientķfica actual
1.Tratamientos hormonales
No se conoce el efecto del tratamiento sustitutivo de los déficits de hormonas sexuales en la población de diįlisis ni de los nuevos moduladores de los receptores de estrógenos como el Raloxifeno (57). En la actualidad se estį llevando a cabo un estudio multicéntrico espańol que sin duda clarificarį aspectos de esta cuestión.
2. PTH humana recombinante
En la actualidad estį emergiendo cómo potencial tratamiento anabólico en la osteoporosis la administración de PTH (1-34) recombinante humana y del anįlogo de la proteķna relacionada con la PTH (PTHrp) humana (60). Se ha demostrado que aumentan la masa ósea en animales de experimentación y humanos (61). La administración subcutįnea de PTH (1-34) humana también ha demostrado su eficacia en pacientes con hipoparatiroidismo (62). No existen datos sobre su comportamiento en pacientes con IRC e "hipoparatiroidismo relativo" pero es probable que en el futuro dispongamos de esta nueva alternativa terapéutica .
3.Conocimiento y posible utilización terapéutica de factores estimuladores o neutralización de factores inhibidores locales
Son numerosos los factores supresores de la formación ósea osteoblįstica. Entre ellos, las IL-11 y IL-4 inducidas por la diįlisis y la "osteogenic protein 1" o "bone morfogenenetic protein 7" producida por las células renales ( y por tanto probablemente ausente en la insuficiencia renal). Por otro lado, los factores activadores osteoblįsticos podrķan jugar un papel en el arsenal terapéutico del futuro (63).
4. Conocimiento de factores genéticos asociados con la osteoporosis y el "hipoparatiroidismo relativo" en pacientes con IRC.
Aunque existe variabilidad entre los resultados y es evidente que la osteoporosis y el "hipoparatiroidismo relativo" en pacientes con IRC estan influidos genéticamente. Conocer estos factores de riesgo nos ayudarį en el futuro a plantear precozmente las estrategias terapéuticas.
BIBLIOGRAFĶA
1. Cannata Andķa JB. Hypokinetic azotemic osteodistrophy. Kidney Int 54: 1000-1016; 1998.
2. Block GA, Hulbert-Shearon TE, Levin NW, Port FK. Association of rerum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 31:607-617; 1998.
3. Fernįndez E, Belart M, Montoliu J. Succesful treatment of massive uremic tumoral calcinosis with daily hemodialysis and very low calcium dialysate. Nephrol Dial Transplant 4: 1207-1209; 1994.
4. Fernįndez E, Torregrosa V, Zįrraga S, Campistol JM. Calcinosis tumoral y calcificaciones vasculares en el trasplantado renal. Nefrologķa 18: 77-83; 1998.
5. Parfitt AM, The hyperparathyroidism of chronic renal failure: A disorder of growth. Kidney Int 52: 3-9; 1997.
6. Hsu CH. Are we mismanaging calcium and phosphate metabolism in renal failure?. Am J Kidney Dis 29: 641-649; 1997.
7. Ramirez JA, Emmett M, White MG, Fathi N, Santa AC, Morawski SG, Fordtran JS. The absorption of dietary phosphorus and calcium in hemodialysis patients. Kidney Int 30: 753-759; 1986.
8. Rufino M, Bonis E, Martķn M, Rebollo S, Martķn B, Miquel R, Cobo M, Hernįndez D, Torres A, Lorenzo V. Is it possible to control hyperphosphatemia with diet, without inducing protein malnutrition?. Nephrol Dial Transplant 13: (Suppl3): 65-67; 1998
9. Campistol JM, Almirall J, Martin E, Torras A, Revert LL. Calcium carbonate-induced calciphylaxis. Nephron 51: 549-550, 1989.
10. Llach F. Calcific uremic arteriolopathy (Calciphylaxis ): An evolving entity?. Am J Kidney Dis 32: 514-518; 1998.
11. Ritz E. Phosphate removal during dialysis- does the membrane matter?. Clinical Nephrol 42: S57-S60; 1994.
12. Hou SH, Zhao J, Ellman CF, Hu J, Griffin Z, Spiegel DM, Bourdeau JE. Calcium and phosphorus fluxes during hemodialysis with low calcium dialysate. Am J Kidney Dis 28: 217-224; 1991.
13. Fernįndez E, Borras M, Pais B, Montoliu J. Low calcium dialysate stimulates parathormone secretion and its long-term use worsens secondary hyperparathyroidism. J Am Soc Nephrol 6: 132-135; 1995.
14. Argiles A, Kerr PG, Canaud B, Flavier JL, Mion C. Calcium kinetics and the long- term effects of lowering dialysate calcium concentration. Kidney Int 43: 630-640; 1993.
15. Fernįndez E, Llach F. Guidlines for dosing of intravenous calcitriol in dialysis patients with hyperparathyroidism. Nephrol Dial Transplant 11: 96-101; 1996.
16. Fukagawa M, Iwasaki Y. Molecular pathogenesis of secondary hyperparathyroidism in renal failure: basic and clinical aspects. Nephrol Dial transplant 14 (Suplpl 1):61-62; 1999.
17. Tominaga Y.Mechanisms of parathyroid tumorigenesis in uremia. Nephrol Dial transplant 14 (Suplpl 1):63-65; 1999.
18. Akizawa T, Ogata H, Koiwa F, Kinusaga E, Ideura T. The role of calcium-sensing receptor abnormality in the pathogenesis of secondary hyperparathyroidism. Nephrol Dial transplant 14 (Suplpl 1):66-67; 1999.
19. Farnebo F, Hoog A, Sandelin K, Larsson C, Farnebo LO. Decreased expression of calcium-sensing receptor messenger ribonucleic acids in parathyroid adenomas. Surgery 124(6): 1094-1098; 1998.
20. Rodriguez M. Direct effect of phosphate on parathyroid function. Nephrol Dial transplant 14 (Suplpl 1):70-72; 1999.
21. Reichel H, Deibert B, Schmidt-Gayk H, Ritz E. Calcium metabolism in early chronic renal failure: implications for the pathogenesis of hyperparathyroidism. Nephrol Dial Transplant 6: 162-169; 1991.
22. Martinez I, Saracho R, Montenegro J , Llach F. A deficit of calcitriol syntesis may not be the initial factor in the pathogenesis of secondary hyperparathyroidism. Nephrol Dial Transplant 11 (Suppl 3): 22-28; 1996.
23. Felsenfeld A, Rodriguez M. Phosphorus, regulation of plasma calcium, secondary hyperparathyroidism: A hypothesis to integrate historical and modern perspective. J Am Soc Nephrol 10: 878-890; 1999.
24. Portale AA, Booth BE, Halloran BE, Morris RC. Effect of dietary phosphorus on circulating concentrations of 1,25-Dihydroxyvitamin D in children with moderate renal insufficiency. J Clin Invest 73: 1580-1589; 1984.
25. Llach F, Massry SG. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J Clin Endocrinol Metab 61: 601-606; 1985.
26. Seidel A, Stein G, Ritz E. Do limited changes in phosphate intake modulate 1,25(OH)2D3 levels in early renal failure?. Clin Nephrol 36: 274-280; 1991.
27. Martinez I, Saracho R, Montenegro J, Llach F. The importance of dietary calcium and phosphorous in the secondary hyperparathyroidism of patients with early renal failure. Am J Kidney Dis 29: 496-502; 1997.
28. Fernįndez E, Fibla J, Betriu A, Piulats JM, Almirall J, Montoliu J. Association between vitamin D receptor gene polymorphism and relative hypoparathyroidism in patients with chronic renal failure. J Am Soc Nephrol 8: 1546-1552; 1997.
29. Marco MP, Martinez I, Fibla J, Amoedo ML, Borrąs M, Saracho R, Almirall J, Fernįndez E. Vitamin D receptor genotypes influences PTH and calcitriol levels in predialysis patients. Kidney Int 56: 1349-1353; 1999.
30. Lopez-Hilker S, Duso AS, Rapp Ns, Martin KJ, Slatopolsky E. Phosphorus restricction reverses hyperparathyroidism in uremia independent of changes in calcium and calcitriol. Am J Physiol 259: F432-F437; 1990.
31. Rodriguez M. Direct effect of phosphate on parathyroid function. Nephrol Dial Transplant 14: 70-72; 1999.
32. Rodriguez M, Martin-Malo A, Martinez ME, Torres A, Felsenfeld AJ, Llach F. Calcemic response to parathyroid hormone in renal failure: role of phosphorus and its effect on calcitriol. Kidney Int 40: 1055-1062; 1991.
33. Bover J, Jara A, Trinidad P, Rodriguez M, Martin-Malo A, Felsenfeld A. The calcemic response to PTH in the rat: effct of elevated PTH levels in uremia. Kidney Int 46: 310-317; 1994.
34. Goodman WG, Ramirez JA, Belin TR, Chon Y, Gales B, Segre GV, Salusky IB. Development of adynamic bone in patients with secondary hyperparathyroidism after intermittent calcitriol therapy. Kidney Int 46: 1160-1166; 1994.
35. Slatopolsky EA, Burke SK, Dillon M A, Renagel Study group. RenagelR, A nonabsorved calcium- and aluminium-free phosphate binder, lowers serum phosphorus and parathyroid hormone. Kidney Int 55: 299-307; 1999.
36. Mucsi I, Hercz G, Uldall R, Ouwendyc M, Francoeur R, Pierratos A. Control of serum phosphate without any phosphate binders in patients treated with nocturnal hemodialysis. Kidney Int 53: 1399-1404; 1998.
37. Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of a extracellular calcium sensing receptor from bovine parathyroid. Nature 366: 575-580; 1993.
38. Wada M, Furuya Y, Sakiyama JI, Kobayashi N, Miyata S Ishii H. The calcimimetic compound NPS-R-508 suppresses parathyroid cell proliferation in rats with renal insuficiency: control of parathyroid cell growth via calcium receptor. J Clin Invest 100: 2977-2983; 1997.
39. Wada M, Ishii H , Furuya Y, Fox J, Nemeth EF, Nagano N, NPS R-568 halts or reverses osteitis fibrosa in uremic rats. Kidney Int 53: 448-453; 1998.
40. Antonsen JE, Sherrrd DJ, Andress DL. A calcimimetic acutely suppresses parathyroid hormone levels in patients with chronic renal failure. Rapid communication. Kidney Int 53 (1): 223-227; 1998.
41. Funahashi H., Tanaka Y., Imai T., Wada M., Tsukamura K., Hayakawa Y., Matsuura n., Kikumori T., Oiwa M., Tominaga Y., Takagi H.. Parathyroid hormone suppression dy 22-oxacalcitriol in the severe parathyroid hyperplasia. J. Endicronol Invest: 21(1):43-7; 1998.
42. Monier-Faugere M.C., Genz Z., Friedler R.M., Qi Q., Kubodera N., Slatopolsky E., Malluche H.H.. 22-Oxacalcitriol suppresses secondary hyperparathyroidism without inducing low bone turnover in dogs with renal failure. Kidney Int Mar; 55(3):821-832; 1999.
43. Hirata M, Katsumata K, Masaki T, Koike N, Endo K, Tsunemi K, Ohkawa H, Kurokawa K, Fukagawa M. 22-Oxacalcitriol ameliorates high-turnover bone and marked fibrosa osteitis in rats with slowly progressive nephritis. Kidney Int 56: 2040-2047; 1999.
44. Kurokawa , Akizawa T, Suzuki M, Akiba T, Ogata E, Slatopolsky E. Effect of 22-oxacalcitriol on hyperparathyroidism of dialysis patients: Results of a preliminary study. Nephrol Dial Transplant 11: 121-124; 1996.
45. Slatopolsky E, Finch J, Ritter C, Denda M, Morrissey J, Brown A, DeLuca H. A new analog of calcitriol , 19-nor-1,25-(OH)2D2, suppresses parathyroid hormone secretion in uremic rats in the absence of hypercalcemia. Am J Kidney Dis 26: 852-860; 1995.
46. Takahashi F, Finch JL, Denda M, Dusso AS, Brown AJ, Slatopolsky E. A new analog of 1,25 (OH)2D3, 19-nor-1,25-(OH)2D2, suppresses serum PTH and parathyroid gland growth in uremic rats without elevation of intestinal vitamin D receptor content. Am J Kidney Dis 30: 105-112; 1997.
47. Martin K.J., Gonzįlez E.A., Gellens M.E., Hamm L.L., Abboud H., Lindberg J.. 19-Nor-1a-25-Dihydroxyvitamin D2 (Paricalcitol) safely and Effectively Reduces the Levels of Intact Parathyroid Hormone in Patients on Hemodialysis. J Am Soc. Nephrol 9: 1427-1432, 1998.
48. Finch JL, Brown AJ, Slatopolsky E. Differential effects of 1,25-Dihydroxy-vitamin D3 and 19-Nor-1,25-Dihydroxy-vitamin D2 on calcium and phosphorus resorption in bone. J Am Soc Nephrol 10: 980-985; 1999.
49. Tan AU, Levine BS, Mazess RB, Kyllo DM, Bishop CW, Knutson JC, Kleinman K.S., Coburn J.W.. Effective suppression of parthyroid hormone by 1a-hydroxy-vitamin D2 in hemodialysis patients with moderate to severe secondary hyperparathyroidism. Kidney Int Vol. 51: 317-323; 1997
50. Stehman-Breen C, Muirhead N, Thorning D, Sherrard D. Secondary hyperparathyroidism complicated by parathyromatosis. Am J Kidney Dis 28: 502-507; 1996.
51. Perez L, Betriu A, Pelayo M, Fernįndez E. New technique of parathyroidectomy to prevent parathyromatosis and hypoparathyroidism. Nephrol dial transplant 14: 1553-1555; 1999.
52. Torres A, Lorenzo V, Hernįndez D, Rodriguez JC, Concepción MT, Rodriguez AP, Hernįndez A, De Bonis E, Darias E, Gonzalez-Posada JM, Losada M, Rufino M, Felsenfeld AJ, Rodriguez M. Bone disease in predialysis, hemodialysis and CAPD patients: Evidence of a better bone response to PTH. Kidney Int 47: 1434-1442; 1995
53. Riggs BL. Vitamin D-receptor genotypes and bone density. N Engl J Med 337: 125-126; 1997
54. Atsumi K, Kushida K, Yamazaki K, Shimizu S, Ohmura A, Inoue T. Risk factors for vertebral fractures in renal osteodystrophy. Am J Kidney Dis 33: 287-293; 1999.
55. Parfitt AM. A structural approach to renal bone disease. J Bone Min Dis 13: 1213-1220; 1998.
56. Stein MS, Packman David K, Ebeling PR, Wark JD, Becker GJ. Prevalence and risk factors for osteopenia in dialysis patients. Am J Kidney Dis 28: 515-522; 1996.
57. Lindberg J, Moe SM. Osteoporosis in end-stage renal disease. Sem Nephrol 19: 115-122; 1999.
58. Kurz P, Monier-Faugere MC, Bognar B, Wernwr E, Roth P, Vlachojannis J, Malluche HH. Evidence for abnormal calcium homeostasis in patients with adynamic bone disease. Kidney Int 46: 855-861; 1994.
59. Weinreich T. Prevention of renal osteodistrophy in peritoneal dialysis. Kidney Int 54: 2226-2233; 1998.
60. Frolik CA, Cain RL, Sato M, Harvey AK, Chandrasekhar S, Black EC, Tashjian AH, Hock JM. Comparison of recmbinat human PTH(1-34) (LY333334) with a C-terminally substituted analog of human PTH-related protein (1-34) (RS-66271): In vitro activity and in vivo pharmacological effects in rats. J Bone Miner Res : 163-172; 1999.
61. Lane NE, Sanchez S, Modin GW, Genant HK, Ini E, Arnaud CD. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. J Clin Invest 102: 1627- 1633; 1998.
62. Winer KK, Yanovski JA, Sarani B, Cutler GB Jr. A randomized, cross-over trial of once-daily versus twice-daily parathyroid hormone 1-34 in hypoparathyroidism. J Clin Endocrinol Metab 83: 3480- 3486; 1998.
63. Hruska K. New concepts in renal osteodistrophy. Nephrol Dial Transplant: 2755-2760; 1998.
![]()