
J.Balasubramaniam, M.D, D.M Kidney Care Centre Tirunelveli, Tamilnadu, India
| DISCUSSION BOARD |
Introduction
Over the years, study of renal physiology has tuned and fine-tuned our knowledge on various pathways and mechanisms involved in the functioning of the kidneys. As newer mediators, molecules and receptors are discovered, new theories evolve; some substantiate and some others disprove old notions; only a few promise new avenues for treatment strategies. But not all get translated into useful therapeutic breakthroughs. Nowhere is this phenomenon well exemplified than in the relationship between prostaglandins (PG) and the kidney.
Mechanism of NSAID action
The various inflammatory mediators, Prostaglandins, Thromboxanes and Leucotreines are derived from phospholipids in the cell membrane via the cyclo-oxygenase and lipo-oxygenase pathways.(Figure 1). NSAIDs, as a group, derive their anti-inflammatory capability by inhibiting the cyclo-oxygenase (COX) pathway and hence the synthesis of prostaglandins. Many physiological actions other than inflammation such as maintenance of gastric mucosal integrity and modulation of renal microvascular hemodynamics, renin release, and tubular salt and water reabsorption are also mediated by the prostaglandins. Positive role of prostaglandins in maintaining the GFR in the face of adverse circumstances has been known to us for sometime. Hence, nephrotoxicity due to NSAIDs is rather inevitable.
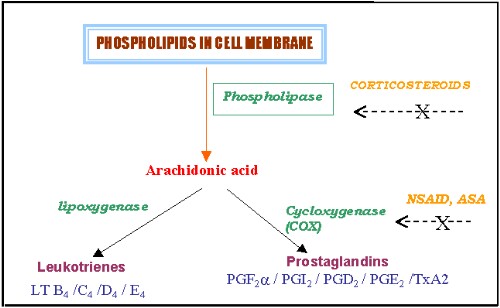
Figure 1
Aspirin had been known for over 100 years, well ahead of our knowledge about role of prostaglandins in renal function1,2,3,4, and its analgesic, anti-inflammatory, antithrombotic actions were recognized at various times. By 1938, its gastritis producing nature was well recognized and was distressing to the physicians. In the 1960s, non-aspirin NSAIDs surfaced but they failed to be "safer aspirins". It was getting clear by the 1970s that inhibition of prostaglandin synthesis via a cyclo-oxygenase (COX) enzyme was central to both the therapeutic and toxic effects of aspirin and non-aspirin NSAIDs. In the 1980s, the clinical consequences of nephrotoxicity, induced by NSAIDs, were abundantly documented.5,6. NSAIDs were recognized to induce edema, hypertension, acute renal failure, and, rarely, the nephrotic syndrome.
Cyclo-oxygenase is twosome…COX 1 and COX 2!
To the delight of many, in the 1990s, it was recognized that there were two enzymes COX 1 and COX 2 involved in the cyclo-oxygenase pathway and there arose the possibility of dissociating the good from the bad effects of NSAIDs.7,8(Figure 2)

Figure 2
Two related isoforms of the COX enzyme have been described9,10 - COX-1 (PGHS-1) and COX-2 (PGHS-2). They possess 60 percent homology in those amino acid sequences apparently conserved for catalysis of arachidonic acid.11-15 The most important differences between the two isoforms are the regulation and expression of the enzymes in various tissues:
Need for Selective COX 2 inhibitors
The differences in the effectiveness with which a particular NSAID inhibits an isoform of cyclooxygenase may affect both its activity and toxicity. It has been proposed that the perfect NSAID would inhibit the inducible COX-2 isoform (thereby decreasing inflammation) without having any effect on the constitutive COX l isoform (thereby minimizing toxicity). Such an agent would maximize effectiveness, without inducing toxicity, particularly gastroduodenal erosions and nephrotoxicity.
Most traditional NSAIDs are nonselective inhibitors of both COX 1 and COX 2. Nimesulide, a selective COX-2 inhibitor, celecoxib, and rofecoxib, two highly selective COX-2 inhibitors, have been in use for sometime now. They have at least a 200 to 300 fold selectivity for inhibition of COX-2 over COX 1. These agents provide analgesia comparable to the nonspecific NSAIDs among patients with rheumatoid arthritis and osteoarthritis and claimed to have several unique benefits, particularly a marked reduction in gastroduodenal toxicity (since COX 1 is involved in gastric cytoprotection) and renal safety. In addition, COX 2 inhibitors have been found to reduce the risk of colonic malignancy. Thus far highly selective COX-2 inhibitors have been extensively used in clinical practice and studies, conferring important benefits with regard to gastrointestinal side effects. But renal safety of these compounds has not been the focus of interest in the available large clinical trials.37
Are COX 2 inhibitors nephrotoxic?
Reports of renal toxicity due to selective COX 2 inhibitors nimesulide, celecoxib, and rofecoxib, are appearing at regular intervals, notwithstanding the initial claims. Nimesulide, a selective COX 2 inhibitor, induced acute renal failure have been known since 1988. In a randomized controlled trial of rofecoxib in elderly persons receiving low salt diet, GFR reduction was as much as caused by non selective NSAID, indomethacin.36
The report by Perazella and Eras18 cautions us about the renal safety of the highly selective COX 2 inhibitors. Celecoxib and rofecoxib induced transient deterioration of renal function in three patients who were from 63 to 73 years of age.19 Notably, these patients were chronically ill with multi-system diseases, including hypertension, arteriosclerotic cardiovascular disease, heart failure, diabetes mellitus, and chronic renal failure. More and more literature 39,40,41 on the renal toxicity of COX 2 inhibitors is accumulating, suggesting a pattern of nephrotoxicity similar to traditional non-steroidal anti-inflammatory drugs. COX 2 inhibitors have also been shown to have adverse effect on blood pressure control in stable hypertensive patients.42 These disturbing reports call for rethinking and renewed look at the renal physiology especially with respect to COX 2, Prostaglandins and Angiotensin II, to get more insight.
Role of A II, Prostaglandins and COX-2 in renal physiology
Angiotensin II(A II) is the most important effector of body sodium and blood pressure maintenance. Control of renin secretion is the rate-limiting step in the formation of A II. Prostaglandins(PG) as paracrine mediators, are believed to be important factors that can alter rennin secretion.(Figure 3).
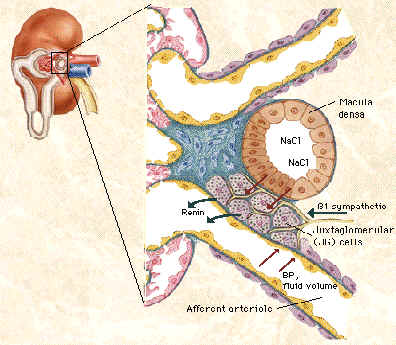
The converse relation, namely, PG stimulating A II release is also true. The macula densa mechanism for the control of renin release has been well established and direct evidence exists for PG dependence of low Nacl- stimulation of renin secretion. But all the while COX protein presence had been established by immunohistology only in cells (vascular endothelial cells, epithelial cells of the Bowman’s capsule and the mesangial cells) away from the granular cells, which secrete renin and there was a gap in our understanduing.
COX-2 receptor in Macula densa
The demonstration of COX 2 in the macula densa and the Thick Ascending Limb (TAL), by Harris et al35 was a breakthrough finding, since it immediately suggested a pathway along which the PG’s produced in the Nacl sensing epithelium could interact with the renin producing granular cells. Now it is very clear that renin secretion and renin gene expression induced by a low-salt-diet and frusemide treatment are mediated partially or completely by COX-2-derived renal prostaglandins. Since the control of renin release is also influenced by other factors than the macula densa, the overall effect of COX 2 inhibition on renin release, A II production and the renal function would depend on the clinical setting. Significant reduction in renin- A II secretion and renal function by COX 2 inhibitors can be expected in conditions in which low Nacl by the macula densa is the major contributing cause for the high reninemic state. The use of diuretics, CCF, cirrhosis, nephrotic syndrome and Bartter’s syndrome are best examples of situations of hyperreninemic state in which renin production is mainly driven by TAL/MD COX 2 and is therefore sensitive to COX 2 suppression.
Now going back to the reports of renal toxicity by COX 2 inhibitors, we find that in most instances, there was a setting of high renin state due to diuretics, cirrhosis, CCF, nephritic syndrome, underlying renal failure or old age.
COX 2 inhibitors and neonatal renal failure
The other group of startling reports was in the case of neonatal renal failure caused by maternal ingestion of COX 2 inhibitors. First it was perinatal vasoconstrictive reversible renal insufficiency associated with maternal nimesulide use.38 Then came the case reports of irreversible renal failure following maternal ingestion of nimesulide by Balasubramaniam.J.3,20 and Licia Peruzi et al.9 Maturation arrest of the tubules and the presence of immature fetal glomeruli were well documented in the renal biopsy of the neonate.20 (Figures 4,5). Interestingly the maternal ingestion had taken place in the last part of the pregnancy suggesting the role of COX 2 in the renal development and maturation occurring in the last part of pregnancy and the early neonatal period.
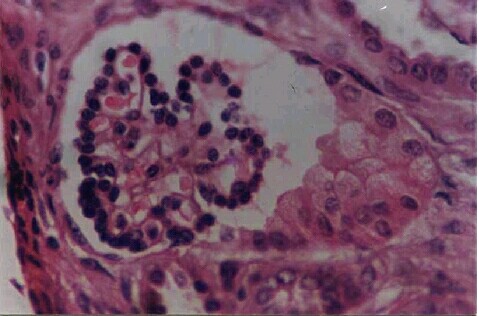
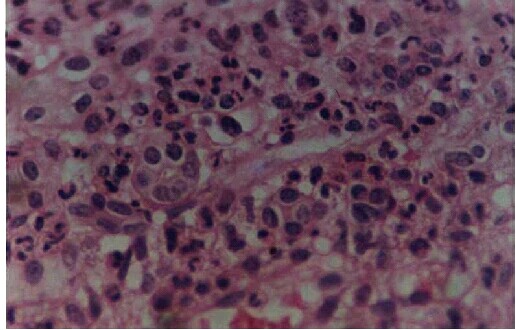
Figure 5. Tubules in various stages of maturation - some with distinct lumina lined by tall columnar cells with eosinophilic cytoplasm and centrally placed nuclei. Others, solid clusters of cells with ill defined lumina and high nucleo-cytoplasmic ratio
Role of COX 2 in renal development:
Fetal and early postnatal kidneys possess functional COX activity and are a rich source of prostaglandins.23,24 Studies have localized COX-2 mRNA and immunoreactive protein in the kidneys of normal rats and determined that renal COX-2 expression is highly developmentally regulated.25 During kidney development, immunoreactive COX-2 is first observed in mid-gestation embryonic stages, notably in cells undergoing induction and/or morphogenesis and for the duration of nephrogenesis (through postnatal wk 2 in the rat. The mature intense form of immunoreactive COX-2 appears primarily in functional nephrons as they mature.25 In the postnatal kidney, COX-2 expression is relatively low at birth, increases in the first two postnatal weeks, and gradually declines to low levels in normal adult rats.25
This expression pattern of COX-2 in the developing kidney is of interest because of the evidence that COX metabolites play important functional and developmental roles in the fetal kidney. There also is evidence that COX metabolites may mediate normal renal development. Chronic administration of indomethacin to pregnant Rhesus monkeys led to renal hypoplasia in the neonates, with kidney mass reduced by 15% compared with control animals.29 The observed defect was specific for the kidney, because in the treated animals, development of other organs was not affected, except for hepatic hypertrophy.
Targeted disruption of murine COX-2 has indicated an important role for this enzyme in renal development.32,33. At maturity in homozygous COX-2 null mice, the kidneys are small, with fewer developed nephrons than in wild-type kidneys. Undeveloped mesenchymal tissue, immature glomeruli, and dysplastic tubules were present in the outer cortex. No apparent developmental or functional abnormalities have been described in mice with targeted disruption of COX-1.34 Of interest, maternal administration of a selective COX-2 inhibitor to wild-type mice and rats during the fetal and/or perinatal period led to renal lesions similar to that observed in the homozygous COX-2 null mice.26
Chronic use of COX inhibitors during human pregnancy has also been related to fetal renal maldevelopment; kidneys from infants who came to term or died in the early postnatal period had few differentiated proximal tubules in the inner cortex and crowding of the glomeruli.30,31 The outer cortex was more severely affected, with evidence of poorly differentiated glomeruli, undifferentiated tubule epithelia, and tubular dilation.
The above hypothesis and the elegant experimental evidences on the role of COX 2 in the renal development have been proved right by the reports of neonatal renal failure following maternal ingestion of selective COX 2 inhibitors during the last stages of pregnancy.20,21 In humans, an increased incidence of oligohydramnios has been observed in women who consumed significant amounts of aspirin, non-selective COX inhibitors or selectiver COX 2 inhibitors during the third trimester of pregnancy.20,21,28 Because the fetal urine is the source of a significant amount of the amniotic fluid, these studies suggested that inhibition of COX led to the suppression of fetal renal function. Most physicians are careful in using/avoiding drugs only during the early pregnancy for the fear of developmental problems to the fetus. This impressive array of experimental and clinical evidences should put us all on guard as irreversible renal failure can result in the newborn unlike the acute renal failure in adults due to COX 2 inhibitors.
Other roles of COX 2:
Renal COX-2 is up-regulated in a variety of experimental nephritis and ablation models and in human lupus nephritis, and COX-2 inhibitors were beneficial in some (but not all) studies.43 Methotrexate38 and corticosteroids may be acting through COX 2 inhibition when used for rheumatic diseases. These issues clearly need more study.
Conclusion :
In summary,
References:
1. Lee JB, Crowshaw K, Takman BH, Attrep KA: The identification of prostag-landins E2, F2a and A2 from rabbit kidney medulla. Biochem J 105:1251-1260, 1967
2. Daniels EG, Hinman JW, Leach BE, Muirhead EE: Identification of prostaglandin E2 as the principal vasodepressor lipid of rabbit renal medulla. Nature 215:1298-1299, 1967
3. Vane JR: Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nature (London) 231:232-235, 1971
4. Dunn MD, Hood VL: Prostaglandins and the kidney: An editorial review. Am J Physiol 233:169-184, 1977
5. Dunn MJ, Zambraski EJ: Renal effects of drugs that inhibit prostaglandin synthesis. Kidney Int 18:609-622, 1980
6. Dunn MJ: Nonsteroidal anti-inflammatroy drugs and renal function. Annu Rev Med 35:411-428, 1984
7. Herschman HR: Prostaglandin synthase 2. Biochim Biophys Acta 1299:125-140, 1996
8. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van de Putte LB, Lipsky PE: Cyclooxygenase in biology and disease. FASEB J 12:1063-1073, 1998
9. Shimokawa, T, Smith, WL. Prostaglandin endoperoxide synthase: The aspirin acetylation region. J Biol Chem 267:12387. (7), 1992
10. Shimokawa, T, Smith, WL. Essential histidines of prostaglandin endoperoxide synthase. His-309 is involved in heme binding. J Biol Chem 266:6168. (8), 1991
11. Shimokawa, T, Kulmacz, RJ, DeWitt, DL, Smith, WL. Tyrosine 385 of prostaglandin endoperoxide synthase is required for cyclooxygenase catalysis. J Biol Chem 265:20073. (9), 1990
12. Toh, H. Prostaglandin endoperoxide synthase contains an EGF-like domain. FEBS Lett 258:317. (10), 1989
13. Lee, SH, Soyoola, E, Chanmugam, P, et al. Selective expression of mitogen-inducible cyclooxygenase in macrophages stimulated with lipopolysaccharide. J Biol Chem 267:25934. (11), 1992
14. Dubois, RN, Abramson, SB, Crofford, L, et al. Cyclooxygenase in biology and disease. FASEB J 12:1063. (12), 1998
15. Sheng, H, Shao, J, Kirkland, SC, et al. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J Clin Invest 99:2254. (26), 1997
16. Tsujii, M, Kawano, S, Du Bois, RN. Cyclooxygenase-2 expression in human colon cancer cells increases metastatic potential. Proc Natl Acad Sci U S A 94:3336. (28), 1997
17. Sheehan, KM, Sheahan, K, O'Donoghue, DP, et al. The relationship between cyclooxygenase-2 expression and colorectal cancer. JAMA 282:1254. (29), 1999
18. Perazella MA, Eras J: C0X-2 inhibitors and acute renal failure. Am J Kidney Dis 35:937-940, 2000
19. Swan, SK, Rudy, DW, Lasseter, KC, et al. Effect of cyclooxygenase-2 inhibition on renal function in elderly persons receiving a low-salt diet. A randomized, controlled trial. Ann Intern Med 133:1, 2000
20. Balasubramaniam J. Nimesulide and neonatal renal failure. Lancet. 12;355(9203):575, 2000
21. Peruzzi L, Gianoglio B, Porcellini MG, Coppo R. Neonatal end-stage renal failure associated with maternal ingestion of cyclo-oxygenase-type-1 selective inhibitor nimesulide as tocolytic. Lancet. 6;354(9190):1615, 1999 22. Balasubramaniam J. Selective COX-2 inhibitors and nephrotoxicity. Am J Kidney Dis. 36(3):675-6, 2000
23. Day NA, Attallah AA, Lee JB: Presence of prostaglandin A and F in fetal kidney [Letter]. Prostaglandins 5: 491-493,1974
24. Moel DI, Cohn RA, Penning J: Renal prostaglandin E2 synthesis and degradation in the developing rat. Biol Neonat 48 : 292-298,1985
25. Zhang MZ, Wang JL, Cheng HF, Harris RC, McKanna JA: Cyclooxygenase-2 in rat nephron development. Am J Physiol 273:F994 -F1002, 1997
26. Komhoff M, Wang JL, Cheng HF, Langenbach R, McKanna JA, Harris RC, Breyer MD: Cyclooxygenase-2-selective inhibitors impair glomerulogenesis and renal cortical development. Kidney Int 57 : 414-422,2000
27. Matson JR, Stokes JB, Robillard JE: Effects of inhibition of prostaglandin synthesis on fetal renal function. Kidney Int 20: 621-627,1981
28. Schoenfeld A, Bar Y, Merlob P, Ovadia Y: NSAIDs: Maternal and fetal considerations. Am J Reprod Immunol 28 : 141-147,1992
29. Novy MJ: Effects of indomethacin on labor, fetal oxygenation, and fetal development in rhesus monkeys. Adv Prostaglandin Thromboxane Res 4: 285-300,1978
30. Kaplan BS, Restaino I, Raval DS, Gottlieb RP, Bernstein J: Renal failure in the neonate associated with in utero exposure to non-steroidal anti-inflammatory agents [Comment]. Pediatr Nephrol 8 : 700-704,1994
31. Voyer LE, Drut R, Mendez JH: Fetal renal maldevelopment with oligohydra-mnios following maternal use of piroxicam. Pediatr Nephrol 8:592 -594, 1994
32. Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA, et al.: Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83:473 -482, 1995
33. Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM, et al.: Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature 378:406 -409, 1995
34. Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, et al.: Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 83:483 -492, 1995
35. Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD: Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest 94 : 2504-2510,1994
36. Suzanne K. Swan et al. : Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. Ann Intern Med. 133:1-9,2000
37. Stubanus M, Riegger GAJ, Kammerl MC, Fischereder M, Krämer BK. Renal side effects of cyclooxgenase-type-2 inhibitor use. Lancet2000; 355: 753
37. S. B. V. Mello, D. M. Barros, A. S. F. Silva, I. M. M. Laurindo and G. S. Novaes: Methotrexate as a preferential cyclooxygenase 2 inhibitor in whole blood of patients with rheumatoid arthritis. Rheumatology 39: 533-536, 2000
38. Landau D, Shelef I, Polacheck H, Marks K, Holcberg G. : Perinatal vasoconstrictive renal insufficiency associated with maternal nimesulide use. Am J Perinatol 16(9):441-444, 1999
39. Zhao SZ, Reynolds MW, Lejkowith J, Whelton A, Arellano FM : A comparison f renal-related adverse drug reactions between rofecoxib and celecoxib, based on the World Health Organization/Uppsala Monitoring Centre safety database. Clin Ther. 2001 Sep;23(9):1478-91.
40. Rocha JL, Fernandez-Alonso J :Acute tubulointerstitial nephritis associated with the selective COX-2 enzyme inhibitor, rofecoxib. Lancet. 16:357(9272):1946-7, 2001.
41. Perazella MA, Tray K: Selective cyclooxygenase-2 inhibitors: a pattern of nephrotoxicity similar to traditional nonsteroidal anti-inflammatory drugs.>Am J Med. 111(1):64-7, 2001
42. John W. Graves, MD, Ingeborg A. Hunder, RN: Mayo Clinic and Mayo Foundation, Rochester, MN: Worsening of Hypertension by Cyclo-oxygenase-2 Inhibitors. J Clin Hypertens 2(6):396-398, 2000.
43. Bernhard K. Krämer : Cyclo-oxygenase-2 and renal function. Nephrol Dial Transplant 16: 180-183, 2001