
Strategies to Retard Progression of Chronic Kidney Disease
Malvinder S. Parmar MD, FRCPC, FACP
E-mail: atbeat@ntl.sympatico.ca
Director of Dialysis. Director, Medical Program (Internal Medicine). Timmins and District Hospital
Timmins, Ontario. Canada.
| DISCUSSION BOARD |
Introduction: The number of patients suffering from end-stage renal disease (ESRD) is continuously growing worldwide with prevalence rates of approximately 412 per million in Canada1 and 975 per million in US2. ESRD represents the tip of the iceberg, where chronic renal insufficiency (CRI), the predecessor of ESRD is a significant and growing problem worldwide. Third National Health and Nutrition Examination Survey (NHANES III)3 from 1988 through 1994 estimated that 800,000 Americans have creatinine levels 176 m
mol/L (³
2.0 mg/dL), and an astonishing 10.9 million people have levels ³
132 m
mol/L (1.5 mg/dL), while there were about 350,000 receiving dialysis. Currently, it is unclear as to what proportion of patients with abnormal serum creatinine will progress to ESRD. Chronic renal insufficiency is associated with an extensive and complex set of physiologic consequences that can result in a number irreversible, but preventable, complications. CRI leads to high incidence of anemia, cardiovascular disease (CVD) [left ventricular hypertrophy (LVH), congestive heart failure (CHF)], metabolic bone disease, and malnutrition in patients reaching ESRD. Many of these conditions have their roots early in chronic kidney disease (CKD), greatly increasing the risk of morbidity and mortality during the course of disease progression4-6. Despite increasing attention to modifiable risk factors, the mortality rate among ESRD patients is 10-20 times higher than the general population. The survival rate of patients on dialysis is 79.7% at 1-year, 65.6% at 2-years, 29% at 5-years and only 8.4% at 10-years2. Hence, the focus, in recent years, has shifted to optimizing care before the initiation of RRT for ESRD (pre-ESRD phase) or more importantly during the phase of chronic kidney disease. It represents a critical period in the evolution of kidney disease and provides opportunity to intervene at an earlier stage to improve overall morbidity and mortality. Diagnosis: CRI is usually irreversible and progressive process that results in ESRD. There is no fixed and widely accepted definition for diagnosis of CRI at present2,3,7. US National Institutes of Health in a 1994 Consensus Statement recommended referral to a nephrologist for patients with serum creatinine level ³
132 :
mol/L (1.5 mg/dL) for females and ³
176 :
mol/L (2.0 mg/dL) for males4. More recently, data have suggested that chronic renal insufficiency (CRI) may be present with only minimal alterations of serum creatinine, >105 :
mol/L (1.2 mg/dL) in woman and >132 :
mol/L (1.5 mg/dL) in men8. Serum creatinine is the most commonly used biochemical parameter for estimating glomerular filtration rate (GFR)9. However, this is often a poor predictor of GFR, as it may be influenced in unpredictable ways by assay techniques, endogenous and exogenous substances, renal tubular handling of creatinine, and other factors (age, sex, body weight, muscle mass, diet, drugs etc)10. GFR is the current "gold standard" for determining the status of kidney function, particularly in patients with kidney failure10. Cockcroft-Gault formula for estimation of GFR (Table 1) has been used for many years that incorporate simple parameters of age, weight and serum creatinine values to derive clearance11. More recently, an algorithm developed in Modification of Diet in Renal Disease (MDRD) study has become widely accepted12 but its usefulness in clinical practice is limited because of its complexity. Table 1: Methods for estimating GFR in ml/min/1.73 m2.
MDRD method:12 Stages of chronic kidney disease: Chronic kidney disease is often progressive even if the original insult has resolved, once GFR falls below approximately 25% of normal. In chronic kidney disease, after there is sufficient renal damage to raise serum creatinine to 132 to 176 m
mol/L (1.5 to 2.0 mg/dL), the GFR declines in majority (85%) of patients by an average rate of 4 ml/min/year13. Following is a suggested classification (Table 2, Figure 1) of the various, although arbitrary, stages of renal dysfunction based on GFR (ml/min/1.73 m2).
Cockcroft-Gault Method:11
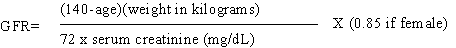
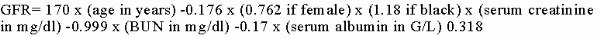
Table 2: Stages of Renal dysfunction:
|
Stage (Renal dysfunction) |
GFR (ml/min/1.73 m2) |
Metabolic Consequences |
|
Normal renal function – Persons at increased risk or with early renal damage |
>90 |
|
|
Mild renal insufficiency (ERI) |
60-89* |
PTH levels start to rise (GFR ~ 60-80) |
|
Moderate renal insufficiency (CRI) |
30-59 |
Decrease in Calcium absorption (GFR <50) Lipoprotein activity falls. Malnutrition. Onset of LVH. |
|
Severe renal insufficiency (Pre-ESRD) |
15-29 |
Triglyceride levels start to rise. Onset of Anemia (EPO deficiency). Hyperphosphatemia. Metabolic acidosis. Hyperkalemia tendency. |
|
ESRD (Uremia) |
<15 |
Azotemia develops. |
* may be normal for age
Figure1: Renal Disease Continuum.
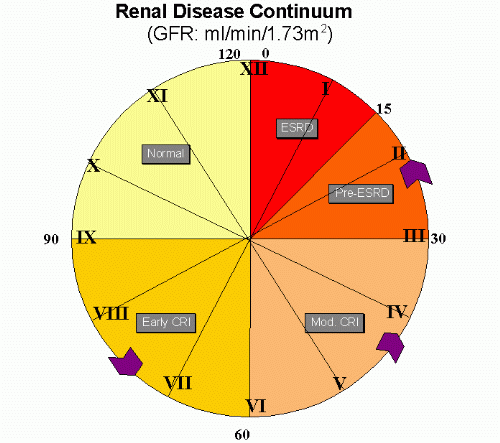
Even when renal function is normal or slightly abnormal, significant renal disease might be present, in high-risk patients or patients with incipient disease (for example, microalbuminuria, diabetic nephropathy, glomerulonephritis, nephrotic syndrome etc.) where early intervention is important to slow this continuum.
Risk Factors for Chronic Kidney Disease:
There is a higher prevalence of common cardiovascular risk factors (Diabetes, hypertension, hyperlipidemia and smoking) in CKD. Diabetes and hypertension are the two most common causes of ESRD and accounts for about 67% of total cases with ESRD. Cardiovascular disease remains the most commonly reported cause of death, representing 40% of all patients1. Early identification and effective control of these common risk factors is of paramount importance for improving both renal and patient outcomes.
The various risk factors (Table 3) can be subdivided in to:
|
Susceptibility Factors |
Initiation Factors |
Progression factors |
|
Increase susceptibility of individual to kidney damage |
Initiate kidney damage |
Cause progressive decline in renal function after initiation of kidney damage |
|
Age Family history of kidney disease Renal transplant |
Diabetes mellitus* Hypertension* Autoimmune diseases Systemic infections Nephrotoxic agents |
Persistent activity of underlying disease Persistent Proteinuria Elevated blood pressure Poor glycemic control Diet: high protein, salt, phosphate diet Hyperphosphatemia Hyperlipidemia* Metabolic acidosis Anemia Cardiovascular disease Smoking* |
*Common Modifiable Cardiovascular Risk factors
Pathogenesis of Progressive Renal Injury:
Pathogenesis of progressive renal injury is complex and multi-factorial (Figure 2) and the current understanding is mainly based on experimental animal models. CKD often progresses by "common pathway" mechanisms, irrespective of the initiating insult14,15.
Early studies focused on functional and structural glomerular changes (glomerular hyperperfusion, hypertension, hyperfiltration, hypertrophy, endothelial cell damage, increased mesangial traffic of macromolecules and proteinuria). Recently there has been increased interest in tubulointerstitial changes as a major determinant of progressive renal injury, especially the role of increased tubular metabolism, ammoniagenesis, generation of free radicals, and of calcium phosphate precipitation in the urinary space and interstitium.
In animal models, a reduction in nephron mass exposes the remaining nephrons to adaptive hemodynamic changes that sustain renal function initially but may be detrimental in the long-term16,17. High glomerular capillary pressure impairs glomerular permeability to proteins, which are then filtered in excess amounts and reach the lumen of the proximal tubule18. Recent studies suggested that proteinuria, previously considered just a marker of the severity of kidney disease, may itself be pathogenic15. In chronic non-diabetic nephropathies, baseline urinary protein excretion rate is the best single predictor of renal disease progression. Higher the urinary protein excretion, faster the subsequent decline in GFR and, even more important, the quicker the progression to ESRD19.

Interventions to Retard Progression of Chronic Kidney Disease:
Primary Prevention: aimed at preventing wide range of acute nephropathies that may initiate progressive loss of renal function by screening high-risk groups, early diagnosis and treatment.
Secondary Intervention: aimed at arresting or significantly retarding the tempo of disease progression once chronic loss of function is established. Interventions designed to interrupt "common pathway" mechanisms of progression of CKD together with targeting coexisting disease modifiers such as hypertension, hyperglycemia and hyperlipidemia.
At each stage of CKD, if there is sudden deterioration of renal function, potentially reversible causes (Table 4) should be evaluated and managed appropriately.
Table 4: Potentially reversible causes of worsening renal dysfunction:
Effective Glycemic and blood pressure control in Diabetes mellitus: Diabetes is a highly prevalent cause of CRI and accounts for a large part of growth of CRI and ESRD over the past decade4,20. Glycemic and blood pressure control blunt its renal complications.
The Diabetes Control and Complications Trial (DCCT) conclusively demonstrated that meticulous control of blood glucose in type 1 diabetes reduced the development of microalbuminuria in 35% of patients21. Similar findings were shown in type 2 diabetes in the United Kingdom Prospective Diabetes Study (UKPDS)22. Other studies in a parallel fashion suggested that glycemic control could reduce the progression of diabetic kidney disease23-26. Similarly, a variety of antihypertensive agents have shown to delay the progression of albuminuria in both type 1 and type 2 diabetes mellitus27. Controlling systemic hypertension is the most important intervention to slow the progression of established diabetic nephropathy28. In diabetic patients with serum creatinine levels over 1.5 mg/dL, treatment with ACE inhibitors produced a 67% reduction in relative risk for progression29. Recently, Angiotensin receptor blockers30-34 have shown their renoprotective effects in both early30 and late31,32 nephropathy due to type 2 diabetes.
Table 5: Management Strategies for Diabetic Nephropathy
Hypertension Control: Hypertension is a well-established cause of chronic kidney disease and a common complication of CRI. The pathophysiology of hypertension is complex and multifaceted. The concurrence of hypertension and kidney disease increases the chance of progression of CRI and risk for complications such as left ventricular hypertrophy (LVH), retinopathy and malignant hypertension36,37.
Hypertension is an important risk factor for progression of CRI irrespective of its cause and effective blood pressure lowering is beneficial in slowing the decline of kidney function 28,38. Best clinical results were observed when adequate blood pressure control was achieved at an early stage of renal insufficiency.
Antihypertensive agents of almost any therapeutic class may be appropriate but Angiotensin-converting enzyme (ACE) inhibitors have been particularly effective in slowing progression of renal insufficiency in patients with and without diabetes mellitus by reducing angiotensin II effects on renal hemodynamics, local growth factors, and perhaps glomerular permselectivity29, 39-42. Non-dihydropyridine calcium channel blockers were also shown to retard progression of renal insufficiency in patients with type 2 diabetes mellitus. Recently, angiotensin receptor blockers (Irbesartan, Valsartan and Losartan) have been shown to have renoprotective effect in diabetic nephropathy and the effect is independent of the reduction in blood pressure30-34. Early detection and effective treatment of hypertension to desired levels is essential to retard the progression of chronic kidney disease. The benefit of aggressive blood pressure control is most pronounced in patients with a urinary protein concentration of >3 g/24-h and benefits patients with both diabetic and nondiabetic renal disease28,43,44.
Table 6: Target Blood Pressure in renal disease45:
Reducing Proteinuria:
Pharmacologic interventions that reduce urinary protein excretion also limit progressive decline in renal function in both diabetic and non-diabetic proteinuric glomerulopathies.
Angiotensin blockade with ACE-inhibitors or angiotensin receptor blockers have clearly shown that at comparable levels of blood pressure control, these agents are more effective than conventional antihypertensive agents in reducing proteinuria, GFR decline and progression to ESRD 30-34,46-48.
Dietary Protein Restriction:
Rationale:
Whether dietary protein restriction slows the progression of CKD remains controversial38,49,50. Various smaller studies suggested that dietary protein restriction might slow the rate of decline in kidney function but were criticized for their small size or methodology. The largest and better-controlled study – the MDRD Study failed to find an effect of protein restriction on decline of kidney function51. Recently, the results of MDRD study have been reexamined using correlational analyses based on achieved protein intake rather than on the intention to treat. Secondary analysis suggested that a lower protein diet (Table 7) retards the progression in both moderate52 and advanced50 renal disease.
Table 7: Dietary Protein Restriction (DPI)
| DPI (g/kg body weight/day)* | When |
| 0.8 g/kg/day | GFR: 25-55 ml/min |
| Nephrotic syndrome | |
| 0.6 g/kg/day | GFR: 13-25 ml/min |
* Assure that >50% of protein is of high biologic value; that there is adequate energy supply (35 kcal/kg/d) and nutritional status is monitored regularly for dietary compliance and prevention of malnutrition.
Dyslipidemia: Lipid abnormalities may be evident with only mild renal impairment and contributes to the progression of CKD53,54 and the increased cardiovascular morbidity and mortality of these patients.
Lipoprotein lipase activity falls in patients with GFR of 50 ml/min or less, and triglyceride levels start to rise when GFR is in the range of 15-30 ml/min55,56. Abnormalities include hypercholesterolemia, elevated ratio of low- to high-density lipoproteins, elevation of lipoprotein(a), and elevated chylomicron remnant levels.
Hypercholesterolemia plays a pathogenic role in the development of progressive renal injury. It is known that LDL binds to the LDL receptors on the surface of the mesangial and epithelial cells and can stimulate cell proliferation, generation of monocyte chemoattractant proteins, and production of matrix proteins53.
Lipid-lowering agents in experimental animals protect against progressive renal injury. In human renal disease, the evidence is limited but preliminary studies57 suggest that these agents may reduce proteinuria and stabilize renal function. HMG-CoA reductase inhibitors block the intracellular production of mevalonate and its metabolites, some of which are critical for the growth and proliferation of cells and thereby may stabilize renal function.
Lp(a) plasma concentration above 30 mg/dL, have been associated with an increased risk of atherosclerotic disease and possibly for progression of CKD. The plasma concentration of Lp(a) can be reduced with antiproteinuric treatment or following renal transplantation. Diet and hypolipidemic therapy are ineffective. In patients with high levels of Lp(a) a very strict control of other atherosclerotic risk factors is important58.
With progression of CRI a generalized disorder of lipid metabolism becomes apparent, irrespective of the underlying cause of kidney disease. Standard regimens to treat dyslipidemia should be employed as per NCEP ATP-III guidelines59. Smoking should be strongly discouraged. Appropriate dose adjustment of various lipid-lowering agents should be made based on level of renal function.
Fibrates increase lipoprotein lipase activity. Nicotinic acid inhibits release of VLDL. Gemfibrozil and HMG-CoA reductase inhibitors are most effective in CKD, but they should not be combined because of increased risk of rhabdomyolysis.
Phosphate and PTH control (Preventing hyperphsophatemia and secondary Hyperparathyroidism):
Hyperparathyroidism appears to be one of the earliest manifestations of renal disease as a response to impaired renal function. Increased parathyroid hormone (PTH) levels have been demonstrated when the GFR falls below 60 to 80 ml/min60,61 and minor changes in bones have been demonstrated in patients with a GFR of as high as 60 ml/min62 and 87% of patients had abnormal bone histology with GFR between 20-59 ml/min63.
Progression of CKD occurs from chronic tubulointerstitial inflammation caused by increases in single-nephron filtered load of phosphate, absolute tubular reabsorption of phosphate, and calcium phosphate product in the tubular lumen and by precipitation of calcium phosphate in the tubules and interstitium, facilitated by reduced concentration of citrate in the tubular fluid (Precipitation-calcification hypothesis, Figure 2)64. This hypothesis is supported in experimental animals showing that a high-phosphate diet aggravates CKD, whereas a low-phosphate diet, administration of phosphate binders, and 3-phosphocitrate (an inhibitor of calcium phosphate precipitation) slows progression of CKD.
In a study of 246 human renal biopsies, elevated tissue calcium levels were found to exist early in renal disease. Renal calcium content correlated significantly with serum creatinine and serum phosphorus, but not with serum calcium. Calcium deposits could be identified in renal biopsies from patients with serum creatinine <132 m mol/L (1.5 mg/dL), indicating that renal calcification begins early in the course of kidney disease. The severity of renal calcification was closely related to hyperphosphatemia and Ca x P product. This finding supports the hypothesis that phosphate-mediated renal calcification is an important factor that may influence the rate of kidney disease progression65.
Hence, it is recommended to reduce the exposure of kidney to calcium phosphate precipitation by adequate fluid intake, modest dietary phosphate restriction and administration of phosphate binders, preferably using calcium-free phosphate binders to avoid calcium load66. Dietary phosphorus should be restricted before GFR falls below 40 ml/min, before development of hyperphosphatemia67-69. The use of vitamin D supplements during pre-ESRD phase is controversial because of concern that it may lead to progression of CKD70. Calcitriol should be used with vigilance to prevent development of elevated calcium-phosphate product, hypercalcemia and over-suppression of PTH. Lower doses of Calcitriol 0.25 m g/day is safe and provides adequate suppression of PTH71,72.
Recent study identifies a strong relationship between elevated serum phosphate, Ca x P product, and parathyroid hormone (PTH) and cardiac causes of death, especially deaths from CAD and sudden death, in hemodialysis patients73.
Table 8: Treatment Goals58:
Table 9: Strategies to control Hyperphosphatemia and secondary Hyperparathyroidism:
Control of Acidosis:
As renal function declines, the acid-base balance is maintained by various compensatory mechanisms, of which an increase in the synthesis of ammonia by proximal tubule is the most important. A defective trapping of ammonia in the medulla (as shown in remnant kidney model) poses further demands on proximal tubules to increase synthesis of ammonia and results in an enhanced concentration of ammonia in the renal cortex. High concentration of free-base ammonia in renal cortex can result in complement activation and interstitial inflammation. Later various metabolic consequences of renal acidosis result including bone demineralization, secondary Hyperparathyroidism, increased protein catabolism, insulin resistance and stunted growth74.
To what extent these observations may be relevant to human renal disease is uncertain. Nevertheless, other detrimental effects of acidosis provide sufficient rationale for treating this complication regardless of any beneficial effect on progressive renal injury. At present there are no generally accepted guidelines. A reasonable goal is to maintain normal serum bicarbonate level by administration of calcium carbonate or sodium bicarbonate.
Sodium and Water balance:
Fluid restriction in the remnant kidney model may cause excessive vasopressin release, increased mesangial cell proliferation and matrix accumulation, and progressive glomerulosclerosis. In addition, fluid restriction may increase the risk for calcium phosphate precipitation in the tubules and may contribute to progressive renal injury by this mechanism.
Sodium retention may contribute to progressive renal injury by its effect on systemic blood pressure. In addition, high-sodium diet abolishes the renoprotective effect of ACE-inhibition in the remnant kidney model.
Hence, intake of fluids should be encouraged and intake of sodium should be restricted to <90 meq/24-h in most patients with CKD. In edematous patients, fluid restriction may be required. Patients with type IV renal tubular acidosis (RTA) may require sodium to enhance potassium excretion and management of hyperkalemia.
Smoking Cessation:
In a retrospective German study, smoking was found to be independent risk factors for development of ESRD in men with diagnosed kidney disease75. Smoking, hypertension and vascular disease was a strong predictor of elevated serum creatinine in non-diabetics older than 65 years76. In patients with CKD, smoking has been associated with higher plasma von Willebrand’s factor and triglycerides that are likely to contribute to the increase in cardiovascular morbidity and mortality in these patients. Therefore, cessation of smoking should be strongly recommended to patients with renal disease.
Anemia:
Anemia of CKD is normochromic and normocytic and is invariably present and begins early, when GFR falls below 30-35% of normal. This is primarily due to decreased erythropoietin (EPO) production by the failing kidney77 but other concomitant factors should be considered in the evaluation of anemia in patients with CKD.
Whether anemia accelerates the progression of kidney disease is controversial. In patients with tubulointerstitial disease that is characterized by more severe anemia is noted to have a slower progression of CKD and yet PCKD shows the opposite – faster progression with less severe anemia.
Anemia decreases both oxygen delivery and protection against oxidative stress and may favor tubular obstruction secondary to interstitial fibrosis. Hypoxia and oxidative stress probably stimulate the production of extracellular matrix by fibroblasts, increasing fibroblasts and creating a vicious cycle.
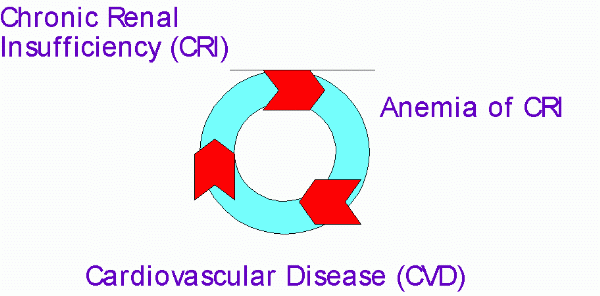
Anemia is independently associated with the development of left ventricular hypertrophy (LVH) and other cardiovascular complications in CRI patients78,79. The CRI, anemia and CVD are associated with each other in a complex vicious cycle and contributes to progression of CKD (Figure 2)
Whether treatment of anemia with recombinant human erythropoietin (rHuEPO) slows the progression of CKD remains elusive. In two small prospective studies 80,81 correcting anemia with rHuEPO significantly slowed the progression of CRI. In another study, it has been shown that rHuEPO plays a role in hypertension, increases arterial vascular resistance bed, decreased venous distensibility. There is little proof that EPO reduces renal oxidative stress or renal cellular apoptosis.
There is a suggestion that treatment of anemia with rHuEPO may slow progression of CKD but further studies are needed to confirm this finding.
The NKF-DOQI guidelines82 recommend that patients with CRI and anemia should be investigated and treated for iron deficiency and other causes of anemia. The maintenance Hct/Hgb target for CRI patients remains uncertain. A target level of 33% to 36% for dialysis and predialysis patients is recommended. Whether this target provides the maximum benefit in term of risk reduction or improvement in quality of life has not been established. Numerous studies in CRI population suggested that this range could be safely achieved with no increase in the rate of progression of CKD83-86. However, treatment of anemia with rHuEPO can elevate blood pressure or require an increase in antihypertensive medications83,85,87 to control hypertension effectively or to prevent its development.
Iron deficiency is common among patients with CRI and is the most frequent cause of failure to respond adequately to rHuEPO. Iron deficiency may develop from chronic blood loss from gastrointestinal tract, blood sampling or other causes. Furthermore, iron stores are depleted rapidly in response to rHuEPO therapy88-89.
The recommendation of transferring saturation of >20% and ferritin >100 ng/ml for dialysis patients could be applied to pre-ESRD patients, as adequate iron stores are necessary to sustain erythropoiesis in response to rHuEPO. Oral iron may be sufficient in the early phases of CRI, but parenteral supplementation may become necessary with advancing CRI, especially if resistance to rHuEPO appears to be developing.