
MUTATIONS IN THE COL4A4 AND COL4A3 GENES CAUSE FAMILIAL BENIGN HEMATURIA
Cèlia Badenas MD. Roser Torra MD
Enfermedades renales hereditarias. Servicio de Nefrología. Fundació Puigvert.
Barcelona. España
E-mail: Roser Torra Balcells
| DISCUSSION BOARD |
INTRODUCTION
Familial benign hematuria (FBH) (MIM 141200) is an autosomal dominant disease. It is characterized by persistent or recurrent microscopic hematuria, not associated with other abnormalities such as renal failure or deafness1. The diagnosis of this benign disease may be difficult to establish, since it is based on a series of negative findings (absence of proteinuria, renal failure or extrarenal symptoms) and the finding of a non-specific ultrastructural lesion, the thin glomerular basement membrane (GBM), and above all on the results of family investigation demonstrating the absence of progression towards renal failure.
Early stages of Alport Syndrome (AS) can be very similar to FBH, both in its clinical features and the electron microscopic appearance.
AS can be inherited as an X-linked, autosomal dominant or autosomal recessive trait2. X-linked forms account for approximately 85% of AS cases and they are due to mutations in the COL4A5 gene. Autosomal dominant and recessive forms account for the rest of cases and they are due to mutations in the COL4A3 and COL4A4 genes. Collagen IV network (formed by COL4A3, A4 and A5 chains) is the major structural component of the GBM of the kidney and these genes are strongly expressed in this tissue3. Some authors have identified mutations in the COL4A3 and COL4A4 genes in families with autosomal recessive AS and hematuria in relatives4-7. However, at least two groups have recently shown that some FBH families are not linked to this locus8,9.
The aim of this study was to test whether FBH could be the result of COL4A3/A4 mutations. We have studied 54 subjects belonging to 11 unrelated FBH Spanish families (Figures 1 and 2). At least one member of each family had undergone a renal biopsy with ultrastructural examination and had a basement membrane thickness below 264 nm. None of the subjects included had either proteinuria or renal failure at the moment of diagnosis, and none of them showed signs of neurosensorial deafness, although no audiogram was systematically performed. We have performed linkage analysis to these families with polymorphic markers flanking the COL4A3/A4 genes and searched for mutations within these genes.
Linkage analysis was performed in all families but two (HFB-4 and HFB-11), which were too small for being tested. In 8 of these 9 families, the results were compatible with linkage to the COL4A3/4 locus (Figure 1). Linkage to COL4A3/4 was excluded in family HFB-3 (Figure 2). As this family included three affected women, linkage analysis to the COL4A5 locus was performed in order to determine whether the 3 affected females could be X-linked AS carriers. Indeed, linkage analysis was compatible with this hypothesis.

Figure 2
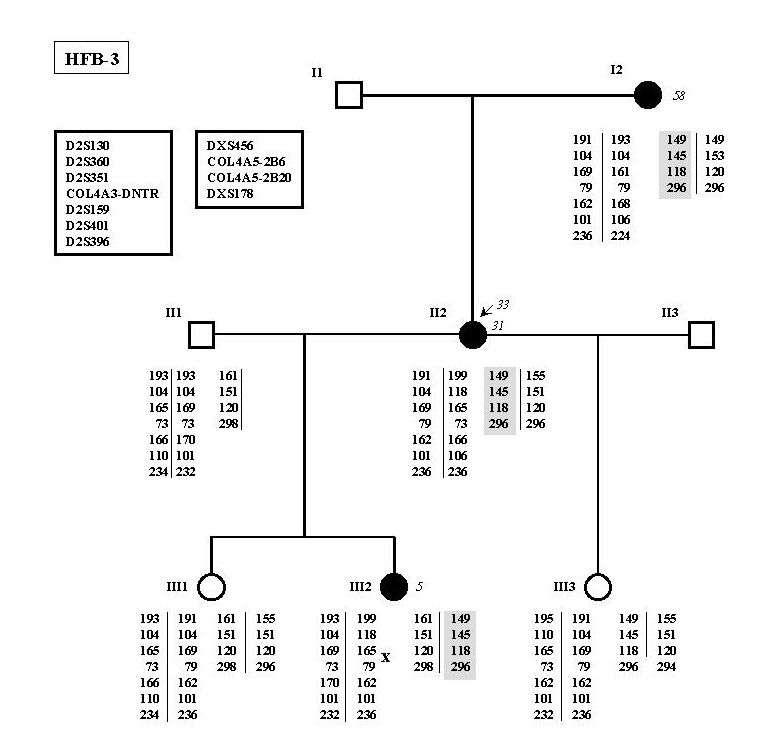
Family HFB-3 in which linkage analysis has been performed for the COL4A3/4 and COL4A5 loci. The identity of each marker is indicated in the box at the upper left. No common haplotype in shared by affected individuals in the COL4A3/4 loci. On the other hand, haplotype of the X-chromosome is shared by the 3 affected women only.
At the moment of diagnosis none of the patients had proteinuria, but during the follow-up period, two patients, both belonging to family HFB-4, developed significant proteinuria without renal failure. HFB-4 seems to have something elseother than FBH.
No mutations were detected in families HFB-3 and HFB-4 as expected if they are not FBH families as suggested by linkage analysis and clinical follow-up.
On the other hand, we have detected 6 mutations in the COL4A3/A4 genes in the 9 remaining families (66.7%). All of them are novel and private (Table 1).
|
Family |
Mutation |
Location |
Predicted effect |
Restriction |
|
|
|
COL4A3 |
COL4A4 |
|
|
site |
|
HFB-1 |
G1015E |
|
Exon 36 |
missense mutation in the collagenous domain |
|
|
HFB-2 |
G985V |
|
Exon 35 |
missense mutation in the collagenous domain |
Rsa Ia |
|
HFB-3 |
|
|
|
|
|
|
HFB-4 |
|
|
|
|
|
|
HFB-5 |
|
|
|
|
|
|
HFB-6 |
|
|
|
|
|
|
HFB-7 |
|
3222insA |
Exon 35 |
frameshift, stop after 1 amino acid |
|
|
HFB-8 |
|
IVS23-1G>C |
Intron 23 |
aberrant splicing of exon 24 |
|
|
HFB-9 |
|
G960R |
Exon 32 |
missense mutation in the collagenous domain |
Msp Ib |
|
HFB-10 |
|
|
|
|
|
|
HFB-11 |
|
31del11 |
Exon 2 |
frameshift, stop after 46 amino acids |
|
Nevertheless, SSCP analysis in the collagen genes is complicated as they are very large genes, no hot spot has been detected and they contain a lot of polymorphisms. As a matter of fact, in the present study 8181.8% of the DNA changes detected by SSCP were polymorphisms (DNA changes not segregating with the disease or found in unaffected subjects). Additionally large rearrangements or mutations in introns or regulatory elements of type IV collagen genes would not be detected by SSCP analysi.
The present study clearly involves theimplicates COL4A4COL4A4 and COL4A3COL4A3 genes in the pathogenesis oif FBH, however, it does not rule out the possibility that other genes expressed in the glomerular basement membrane may be involved in some families with FBH. Possible effects of COLA43 and COL4A4COL4A4 mutations in FBH could be a reduced cross-linking in the type IV collagen network, a decreased content of type 4 collagen novel chains in the GBM and eventually a decreased thickness and stability of the GBM. Why some mutations cause AS while others only cause FBH is still not known, but FBH could be considered as an intermediate phenotype of AS in which a gene-dosage effect is present.
Molecular diagnosis of FBH is still not easily available, as i) COL4A3COL4A/43/4 genes are very large and difficult to screen for mutations, ii) no hot spots have been described up to knowas yet and iii) genetic heterogeneity has been described. The identification of other genes involved in FBH will be of particular interest for clarifying the pathogenesis of this disorder and perhaps for the development of an effective treatment for AS.
REFERENCES