|
Discussion Board
Paneles de Discussión
Paneais de Discussio
Free Papers
Comunicaciones libres
Comunicaçoes livres
Home cin2003
Volver al Inicio cin2003
Voltar ao inicio cin2003
|
Hipertrofia glomerular en la nefropatía diabética
Dra. Ana Maria Cusumano
Sección Nefrología, Departamento de Medicina Interna
CEMIC (Centro de Educación Médica e Investigación Clínica).
Buenos Aires-Argentina
La hipertrofia glomerular es una de las alteraciones estructurales más precoces de la nefropatía diabética, tanto en el tipo 1 como en el tipo 2. La describió por primera vez el médico francés Pierre-Francois-Olivier Rayer en 1839, obviamente en un tipo 2, de la siguiente forma: "En la diabetes se encuentra hipertrofia de la corteza renal...... sin ninguna otra anormalidad excepto hipertrofia.........los vasos se encuentran aumentados de tamańo y los corpúsculos de Malpighi son mucho más prominentes" (1).
La importancia de comprender su fisiopatología reside en que su presencia se asocia al desarrollo de glomeruloesclerosis, y no sólo en la nefropatía diabética. Así se ha comprobado a nivel experimental en modelos de diabetes mellitus, ablación renal, y envejecimiento renal (2-5), y en el humano en glomerulopatías evolutivas como la nefropatía diabética (6), la nefropatia a cambios mínimos con evolución hacia la esclerosis focal y segmentaria (7), la glomerulosclerosis focal y segmentaria y la nefropatía por IgA (8). Basados en estos hechos, se ha sugerido que la presencia de hipertrofia glomerular es uno de los factores no inmunológicos más importantes que contribuyen al desarrollo de glomerulosclerosis.
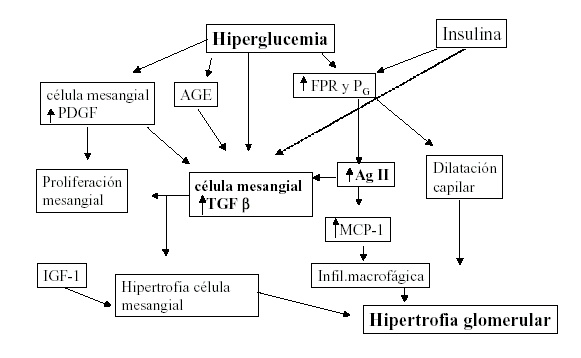
Ahora bien, esta hipertrofia glomerular en la nefropatía diabética se debe a: żun mecanismo adaptativo de aumento del volumen por dilatación de la luz de los capilares, un aumento del número real de células o un incremento del volumen de las mismas? En realidad, la presencia de hiperglucemia desencadena varios mecanismos que confluyen para iniciar los cambios funcionales y estructurales que determinan la hipertrofia glomerular, entre ellos modificación de la hemodinamia corpuscular, y estimulación de la proliferación e hipertrofia celulares.
La hiperglucemia y la hemodinamia glomerular:
La hiperglucemia modifica la hemodinamia glomerular, aumentando el flujo plasmático renal y la presión capilar, gracias a una relativa mayor reducción en la resistencia preglomerular versus la postglomerular (9), probablemente mediada por un aumento en la producción de oxido nítrico a nivel de la arteriola aferente (10). Estas alteraciones hemodinámicas determinan el fenómeno de hiperfiltración, traducido en la clínica por el aumento de la tasa de filtración glomerular.
El incremento de la presión glomerular (PG) provoca la dilatación del capilar; y por ley de Laplace, ante un aumento del radio se genera un incremento en la tensión de la pared. Al transmitirse este aumento de tensión sobre las células endoteliales y mesangiales, se induce la liberación de Angiotensina (Ag) II, que aumenta la expresión del factor de crecimiento transformante 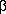 (TGF ), fibrogénico, considerado el nexo entre la hipertensión capilar glomerular y el desarrollo de glomerulosclerosis. (TGF ), fibrogénico, considerado el nexo entre la hipertensión capilar glomerular y el desarrollo de glomerulosclerosis.
La presencia de un sistema renina angiotensina (SRA) intacto a nivel renal es imprescindible para que el aumento de la PG desarrolle injuria glomerular, como lo revelan los estudios de Zatz y colaboradores, que demuestran que el tratamiento con un inhibidor de la enzima de conversión de la angiotensina disminuye la hipertensión capilar glomerular y previene el desarrollo de la nefropatía diabética en ratas, independientemente del control glucémico (11). Estas investigaciones constituyeron la base experimental que generó los numerosos estudios realizados en pacientes diabéticos, que han demostrado categóricamente que la inhibición del SRA, o el bloqueo de los receptores AT 1 mejoran la evolución de la nefropatía diabética.
Además del aumento del tamańo glomerular por la dilatación de los capilares, aumenta el número y longitud de los mismos (12-13), y el consiguiente incremento de la superficie total capilar contribuye aún más al aumento de la filtración glomerular.
Por otro lado, la hipertensión glomerular inducida por Ag II estimula a la célula mesangial a expresar MCP-1 (monocyte chemoattractant protein-1), que también contribuiría a la patogénesis de la nefropatía diabética a través de reclutar macrófagos hacia el glomérulo, donde sirven como una fuente de potentes citokinas proinflamatorias y profibróticas (14).
La hiperglucemia y la proliferación e hiperplasia celulares.
La hiperglucemia induce, a nivel experimental, la proliferación de células glomerulares, tanto in vivo como in vitro (15-16).
Young y colaboradores, marcando con antígeno proliferante nuclear celular (PCNA) en ratas, comprobaron que hay proliferación celular mesangial prominente pero autolimitada, máxima al día 3 post inducción de la diabetes, que vuelve a lo basal después de los 30 días. Esto se asoció con la expresión de novo de  actina, un marcador de activación del fenotipo de la célula mesangial. Se agregó a esto infiltración de macrófagos y linfocitos, máxima al día 30, que contribuyó en menor grado a la proliferación celular. El tratamiento con insulina, en este estudio, previno la proliferación inicial, la expresión de actina y la infiltración macrófago-linfocitaria (16). actina, un marcador de activación del fenotipo de la célula mesangial. Se agregó a esto infiltración de macrófagos y linfocitos, máxima al día 30, que contribuyó en menor grado a la proliferación celular. El tratamiento con insulina, en este estudio, previno la proliferación inicial, la expresión de actina y la infiltración macrófago-linfocitaria (16).
Del mismo modo, células mesangiales en cultivo, tanto de ratón como humanas, expuestas a medio hiperglucémico, exhiben inicialmente una respuesta proliferativa, seguida luego por una etapa antiproliferativa, pero esta última acompańada por hipertrofia celular (17,18).
El responsable de la proliferación inicial sería el factor de crecimiento derivado de las plaquetas (PDGF), que al mismo tiempo estimula la expresión de TGF , y éste sería el responsable de que cese la proliferación, pero simultáneamente provoca la hipertrofia celular (17,19).
Es decir que en la glomerulopatìa diabética ocurrirían tanto proliferación (inicial y autolimitada) como hipertrofia celular (más tardía).
También se ha descripto hipertrofia podocitaria, asociado con un ensanchamiento de los procesos pedicilares, en pacientes con diabetes tipo 2 (20).
Un factor de crecimiento que también juega un rol en la hipertrofia glomerular es el factor de crecimiento símil insulina 1 (IGF-1) (21). Éste media la hipertrofia de células mesangiales a través de una vía dependiente de la calcineurina (22-23), y la administración de un inhibidor de la misma, la Ciclosporina A, evita la hipertrofia glomerular en ratas (24).
Rol del TGF .
La hiperglicemia por sí misma, así como los productos avanzados de glicosilación (AGE), la Ag II inducida por el aumento de tensión de la pared y el PDGF, aumentan la expresión del TGF (16,17,25-28). Éste detiene a las células en la fase G1 del ciclo celular (17), determinando la hipertrofia de las mismas, ya que es en esta etapa cuando incrementan su tamańo y se estimula la síntesis proteíca y de RNA mensajero, previo paso a la siguiente fase G2, de preparación para la mitosis (29)..
Por otro lado, el TGF induciria fibrosis glomerular, aumentando la producción de matriz extracelular (incluyendo colágeno tipo 1, III y IV, fibronectina, laminina, y proteoglicanos (28,32) e inhibiendo la actividad de enzimas que degradan las proteínas de matriz a través de la vía del inhibidor 1 de activación del plasminógeno (PAI-1).
Confirmando el rol del TGF , la administración de anticuerpos neutralizantes para anti-TGF previno el agrandamiento glomerular y suprimió la expresión de genes que codifican para componentes de matriz extracelular en el modelo de ratón diabético insulinodependiente. Similar experiencia, en el ratón obeso con diabetes tipo II, previno casi completamente el aumento en la expresión de colágeno y fibronectina, y la expansión mesangial (30,31).
En síntesis, la hiperglucemia estimula la proliferación y/o hipertrofia de una serie de células a nivel glomerular, que estimulan la síntesis de matriz mesangial, siendo el mediador el TGF (27,28,32). Así como es necesario un SRA intacto, la inducción del TGF , también es necesaria para que se produzca la hipertrofia renal y la acumulación de proteínas de matriz extracelular.
La hiperglucemia y el SRA:
En la diabetes clínica frecuentemente la actividad de renina plasmática está disminuida. Se especula que esto podría deberse a la glicación no enzimática de la prorrenina, con disminución de la conversión a renina activa (33). Sin embargo, la renina plasmática puede no reflejar la situación a nivel renal.
Varios estudios indican la existencia de un SRA local, que se regularía en forma independiente del sistémico, por lo menos a nivel experimental, y que estaría activado en la nefropatía diabética. Así, se ha comprobado que: los receptores glomerulares de Ag II están disminuidos en la rata diabética después de 3-4 semanas de inducida la enfermedad (34); la enzima conversora de la angiotensina se encuentra aumentada a nivel glomerular y vascular (35); hay disminución de los receptores AT 1 en células mesangiales cultivadas en medio hiperglucémico (36) y, los niveles de Ag II a nivel glomerular se encuentran varias veces por encima de los sistémicos (37). Además, la célula mesangial propiamente dicha contiene todos los elementos del SRA, incluyendo renina, angiotensinógeno, receptores AT1, y enzima convertidora de la Ag (38).
La hiperglucemia per se puede aumentar la síntesis de Ag II por las células mesangiales y reducir la degradación de la misma por inhibición enzimática (39), aumentando, por lo tanto los niveles de Ag II. De este modo, actuaría sinérgicamente con la Ag II ya generada localmente por el aumento de PG, estimulando la hipertrofia renal y la síntesis de matriz extracelular. Parte del efecto, como ya se describió, sería a través de la estimulación de la inducción del TGF .
Además, la Ag II incrementa a nivel de la célula mesangial la producción de radicales libres, y el estrés oxidativo consiguiente podría tener un rol en la hipertrofia glomerular, dado que se estimula la kinasa hipertrófica serina-treonina Akt/proteina kinasa B a través de una vía redox-dependiente (40,41).
Rol de la hiperinsulinemia en la diabetes tipo 2.
. La diabetes tipo 2 es la etapa final de un cúmulo de alteraciones metabólicas, que están presentes desde antes que el paciente presente hiperglucemia. Así, a una primera fase con tolerancia normal a la glucosa, pero con resistencia insulínica y distintos grados de hiperinsulinemia, le sigue una fase de intolerancia a la glucosa, con hiperinsulinemia de ayuno persistente (42,43). Las anormalidades funcionales renales comienzan durante esta última fase, específicamente el incremento en la tasa de filtración glomerular (44). No hay estudios en el humano que investiguen si las anormalidades estructurales están presentes en esta fase. Los mecanismos responsables del aumento de la tasa de filtración glomerular en esta etapa se desconocen, y no se ha demostrado que la hiperinsulinemia per se juege un rol.
Nosotros observamos, en monos rhesus (Macaca mulata) obesos y ańosos, un modelo animal que presenta similares anormalidades metabólicas que la diabetes tipo 2 en el humano (45,46), que los individuos hiperinsulinémicos tenían glomérulos de significativamente mayor tamańo comparados con el grupo de monos normales de igual edad. El tamańo glomerular correlacionó significativamente en forma directa con los niveles de insulina plasmática y el peso corporal, no observándose diferencias en el score de glomerulosclerosis o la expansión mesangial. Los monos con diabetes manifiesta también presentaron hipertrofia glomerular, pero en ellos se asoció a mayor glomerulosclerosis, mayor número de glomérulos en oblea y engrosamiento de la membrana basal. Concluimos en ese estudio que la hipertrofia glomerular aparece durante la fase de hiperinsulinemia, siendo el cambio estructural más precoz en este modelo animal, sugiriendo que algún efecto metabólico durante esta fase promueve la hipertrofia glomerular (47).
La hiperinsulinemia se ha propuesto como un mediador de la hipertrofia glomerular, y hay algunas evidencias indirectas que sugieren que jugaría un rol en la aparición de la misma. En el glomérulo hay una alta concentración de receptores de insulina (48), y la estimulación de éstos podría resultar en otros efectos más allá de la captación de glucosa. Así, la infusión aguda de insulina, suficiente para producir hiperinsulinemia, con clamp de glucosa euglucémica, aumenta la presión hidrostática capilar, disminuye la resistencia de la arteriola aferente e incrementa el flujo plasmático renal, resultando en hiperfiltración en la rata no diabética; cambios similares se observan en la rata insulino dependiente que recibe tratamiento crónico con insulina, en dosis suficientes como para prevenir glucosuria (49).
El aumento de PG, como ya se mencionó, provoca la liberación de Ag II, y ésta y la insulina tienen efectos aditivos sobre la expresión del TGF 1 por parte de la célula mesangial (50).
Varios estudios en pacientes han mostrado que la obesidad (presente en la gran mayoría de pacientes con diabetes tipo 2) se asocia a glomerulomegalia, en algunos casos con glomerulosclerosis (51,52). Tanto a nivel experimental como humano se observan similares efectos hemodinámicos en la obesidad que los provocados por la hiperglucemia: aumento del flujo sanguíneo renal y de la tasa de filtración glomerular, con aumento de la fracción de filtración y de la presión hidráulica transcapilar, mediados por vasodilatación de la arteriola aferente (53,54). Nuestro estudio en el mono rhesus no permitió diferenciar la importancia relativa de los niveles de insulina versus el peso corporal de estos animales.
Asimismo, se ha comprobado, a nivel experimental en perros con obesidad temprana que hay proliferación precoz mesangial y endotelial (manifestado con marcación con PCNA). En este mismo modelo, con inmunomarcación se observa aumento en la expresión del TGF 1. Estos animales, similarmente al humano obeso, tienen hiperinsulinismo (dos a tres veces el valor del perro delgado), resistencia insulínica e hiperfiltración glomerular (55).
Además, la euglucemia (ya sea en el contexto de control glucémico estricto por el tratamiento con insulina intensificada o en el trasplante pancreático), puede prevenir, y aún mejorar una vez instalado, el dańo estructural, incluyendo el aumento del volumen glomerular y el engrosamiento de la membrana basal (56-57).
Por otro lado, la presencia de hiperinsulinemia puede ser importante en pacientes que no padecen diabetes. Así, se ha observado una relación directa, lineal, entre los niveles de insulina plasmática y la hipertrofia glomerular en pacientes con nefroesclerosis hipertensiva, lo que aumenta la posibilidad que los niveles de insulina plasmática (o un factor asociado) puedan influenciar la estructura glomerular (58). Además, el aumento de la resistencia periférica a la insulina y la hiperinsulinemia consiguiente son frecuentes en la enfermedad renal no diabética, aparecen tempranamente, y son por lo tanto candidatos que pueden mediar las alteraciones progresivas en la estructura glomerular que caracterizan la insuficiencia renal crónica (59).
Otros estudios a nivel experimental proveen mayor evidencia del vínculo que podría existir entre la insulina plasmática y la hipertrofia glomerular. Así, en la rata obesa Zucker, modelo caracterizado por obesidad, hiperinsulinemia e hiperlipidemia que desarrolla glomerulosclerosis, hay aumento del tamańo glomerular, y el tratamiento con acarbosa, que reduce los niveles de insulina, mejora la glomeruloesclerosis (60). La administración de insulina a ratas no diabéticas altera la composición de la matriz glomerular, y si se agrega la insulina a células mesangiales in vitro, se altera el perfil de las proteínas de la matriz mesangial, con un incremento en el colageno I y III (61,62).
Por lo tanto, no puede descartarse, a la luz de estos hechos que la hiperinsulinemia tenga un rol protagónico en el desarrollo de la hipertrofia glomerular en la diabetes tipo 2. La presencia de hipertrofia glomerular desde la etapa de resistencia insulínica, de confirmarse, tendría importantes implicancias terapéuticas, dado que modificaría el momento de inicio del tratamiento.
Proteína ósea morfogénica 7 (BMP 7).
En la nefropatía diabética, así como en otras injurias renales, se encuentra disminuida la expresión a nivel renal de esta proteína, que se expresa normalmente en la médula renal, el tubo colector cortical y los podocitos. (63,64).
Recientemente, se observó que la administración de BMP-7 a ratas con nefropatía diabética disminuía el aumento de matriz mesangial, prevenía el desarrollo de glomeruloesclerosis y revertía parcialmente la hipertrofia glomerular, normalizando la tasa de filtración glomerular y disminuyendo la proteinuria. Estos efectos ocurrieron sin que se modifiran los niveles de hipertensión arterial (64).
El mecanismo de acción podría ser por inhibición de la seńalización intracelular del TGF-.
Estos resultaron abren la expectativa de una nueva línea de tratamiento de la nefropatía diabética, focalizado en la reparación de la injuria.
Conclusión:
La hipertrofia glomerular es una de las lesiones estructurales más precoces de la nefropatía diabética. Se produce porque la hiperglucemia modifica la hemodinamia glomerular (provocando dilatación del capilar e hiperfiltración), al mismo tiempo que estimula en forma directa la proliferación e hipertrofia celulares, siendo el mediador el TGF . Para que ocurra, es necesario un SRA intacto. En el tipo 2, además, la hiperinsulinemia o un factor asociado presente en la etapa de resistencia insulínica, podría jugar un rol en su fisiopatología. La base del tratamiento consiste en el bloqueo del SRA, a través de la inhibición de la enzima conversora o de antagonizar los receptores AT1 de la Ag.
Bibliografía.
- Rayer P. Traité des maladies des reins et des altérations de la sécrétion urinaire. Etudiées en elles-mêmes et dans leurs rapports avec les maladies de uretères, de la vessie, de la prostate et de l’ urètre. Paris, France. Librairie de l’ Académic Royale de Medécine, NB, 1839.
- Anderson S, Rennke HG, García Dl, Brenner BM. Short and long term effects of antihypertensive therapy in the diabetic rat. Kidney Int 36: 526-536, 1989.
- Daniels BS, Hostetter TH. Adverse effects of growth in the glomerular microcirculation. Am J Physiol 258 (Renal fluid electrol Physiol 27): F1409-1416, 1992.
- Fujihara CK, Limongi DMZP, Oliveira HCF, Zatz R. Absense of focal glomerular sclerosis in aging analbuminemic rats. Am J Physiol 262: R947-R954, 1990
- Floege J, Burns MW, Alpers CE et al. Glomerular cell proliferation and PDGF expression precede glomerulosclerosis in the remnant kidney model. Kidney Int 41: 297-309, 1992.
- Osterby R, Gundersen HJG. Glomerular size and structure in diabetes mellitus. Early abnormalities. Diabetologia 11: 225-229, 1975.
- Fogo A, Hawkins EP, Berry PL, et al. Glomerular hypertrophy in minimal change disease predicts subsequent progression to glomerular sclerosis. Kidney Int 38: 115-123, 1990.
- Thót T, Takebayashi S. Glomerular hypertrophy as a prognostic marker in childhood IgA nephropathy. Nephron 80: 285-291, 1998.
- Hostetter TH, Troy JL, Brenner BM. Glomerular hemodinamics in experimental diabetes mellitus. Kidney Int 19: 410-415, 1981
- Komers R, Allen TJ, Cooper ME. Role of endothelium-derived nitric oxide in the pathogenesis of the renal hemodinamyc changes of experimental diabetes. Diabetes 43: 1190-1197, 1994.
- Zatz R, Dunn BR, Meyer TW, Anderson S, Rennke HG, Brenner BM. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest 1986: 1925-1930, 1986.
- Marcussen N, Nyengaard JR, Christensen S. Compensatory growth of glomeruli is accomplished by an increase number of glomerular capillaries. Lab Invest 70: 868-874, 1994.
- Nyengaard JR, Rasch R. The impact of experimental diabetes mellitus in rats on glomerular capillary number and size. Diabetologia 36: 189-194, 1993.
- Kato S, Luyckx VA, Ots M, et al.Renin angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney Int 56: 1037-1048, 1999.
- Romen W, Takahashi A. Autoradiographic studies on the proliferation of glomerular and tubular cells of the rat kidney in early diabetes. Virchows Arch B 40: 339-345, 1982.
- Young BA, Johnson RJ, Alpers CE et al: Cellular events in the evolution of experimental diabetes nephropathy. Kidney Int 47: 935-344, 1995.
- Wolf G, Sharma K, Chen Y, Ericksen M, Ziyadeh FN. High glucose-induced proliferation in mesangial cells is reverse by autocrine TGF-. Kidney Int 42: 647-656, 1992.
- Nahman NS, Leonhardt KL, Cosio FG, Herbert CL. Effects of high glucose on cellular proliferation and fibronectin production by cultured human mesangial cells. Kidney Int 41: 396-402, 1992.
- Inaba T,Ishibashi S, Gotoda T et al. Enhanced expresión of platelet-derived growth factor receptor by high glucose. Involvement of platelet derived growth factor in diabetic angiopathy. Diabetes 45: 507-512, 1996.
- Pagtalunan ME, Miller PL, Jumping-Eagle S et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest 99: 342-348, 1997.
- Flyvbjerg A, Bornfeldt KE, Marshall SM, Arnqvist Orskov H. Kidney IGF-1 mRNA in initial renal hypertrophy in experimental diabetes in rats. Diabetología 33: 334-338, 1990.
- Hirschberg R, Addler S. Insulin-like growth factor system and the kidney: Physiology, pathophysiology, and therapeutic implications. Am J Kid Dis 31: 901-919, 1998.
- Gooch JL, Tang Y, Ricono JM, Abboud HE. Insulin-like growth factor-I induces renal cell hypertrophy via a calcineurin-dependent mechanism. J Biol Chem 276: 42492-42500, 2001.
- Gooch JL, Barnes JL, García S, Abboud HE. Calcineurin is activated in diabetes and is required for glomerular hypertrophy and ECM accumulation. Am J Physiol 284: F144-F154, 2003.
- Pugliese G, Pricci F, Romeo G et al. Upregulation of mesangial growth factor and extracellular matrix synthesis by advanced glycation end products via receptor-mediated mechanisms. Diabetes 46. 1881-1887, 1997.
- Shankland SJ, Scholey JW. Expression of transforming growth factor-1during diabetic renal hypertrophy. Kidney Int 45: 430-442, 1994.
- Sharma K, Siyadeh FN. Hyperglycemia and diabetic kidney disease: the case for transforming growth factor- as a key mediator. Diabetes 44: 1139-1146, 1995.
- Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-. J Clin Invest 93: 536-542, 1994.
- Shankland SJ, Wolf G. Cell cycle regulatory proteins in renal disease: role in hypertrophy, proliferation and apoptosis. Am J Physiol 278: F 515-529, 2000.
- Sharma K, Yin Yguo K, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 45: 522-530, 1996.
- Ziyadeh FN, Hoffman BB, Han DC, et al. Long term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal anti-transforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97: 8015-8020, 2000.
- Han DC, Isono M, Hoffman BB, Ziyadeh FN. High glucose stimulates proliferation and collagen I type synthesis in renal cortical fibroblasts: mediation by autocrine activation of TGF-beta. JASN 10: 1891-1899, 1999.
- Amemiya S, Ishihara T, Higashida K, Kusano S, Ohyama K, Kato K. Altered synthesis of renin in patients with insulin-dependent diabetes: Plasma prorenin as a marker predicting the evolution of nephropathy. Diabetes Res Clin Pract 10: 15-22, 1990.
- Ballerman BJ, Skorecki KL, Brenner BM. Reduced glomerular angiotensin II receptor density in early untreated diabetes mellitus in the rat. Am J Physiol 247: F110-F116, 1984.
- Anderson S, Jung FF, Inglefinger JR. Renin-angiotensin system in diabetic rats: functional, immunohistochemical and molecular biologic correlations. Am J Physiol 265: F477-F486, 1993.
- Amiri F, Garcia R. Regulation of angiotensin II receptors and PKC isoforms by glucose in rat mesangial cells. Am J Physiol 276: F691-F699, 1999.
- Seikaly MG, Arant BS Jr, Seney FD Jr. Endogenous angiotensin concentrations in specific intrarrenal fluid compartments in the rat. J Clin Invest 86: 1352-1357, 1990.
- Andrade MC, Quinto BM, Carmona AK et al. Purification and characterization of angotensin-I-converting enzimes from mesangial cells in culture. J Hypertension 16: 2036-2074, 1998.
- Singh R, Alavi N, Singh AK, Leehey DJ. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes 48: 2066-2073, 1999.
- Jaimes A, Galceran JM, Raij L. Angiotensin II induces superoxide anion production by mesangial cells. Kidney Int 54: 775-784, 1998.
- Gorin Y, Kim NH, Feliers D et al. Angiotensin II activates Akt/protein kinase B by an arachidonic acid/redox-dependent pathway and independent of phosphoinositide 3-kinase. FASEB J 15: 1909-1920, 2001.
- Beck-Nielsen H, Groop LC. Metabolic and genetic characterization of prediabetic states. J Clin Invest 94: 1714-1721, 1994.
- Kruszynska YT, Olefsky JM. Cellular and molecular mechanisms of non-insulin dependent diabetes mellitus. J invest Med 44: 413-428, 1996.
- Nelson RG, Bennet PH, Beck GJ, et al. Development and progression of renal disease in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetic Renal Disease Study Group. N Engl J Med 335: 1636-1642, 1996.
- Hansen BC, Bodkin NL. Heterogeneity of insulin responses: Phases leading to type 2 (non-insulin-dependent) diabetes mellitus in the rhesus monkey. Diabetología 29: 713-719, 1986.
- Hansen BC: Primate animal models of type 2 diabetes mellitus. In LeRoth D, Taylor S, Olefsky JM (eds): Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia, Lippincott, Williams and Wilkins, 2000, pp 734-743.
- Cusumano AM, Bodkin NL, Hansen B, et al. Glomerular hypertrophy is associated with hyperinsulinemia and precedes overt diabetes in aging rhesus monkeys. Am J Kid Dis 40: 1075-1085, 2002.
- Kurokawa K, Silverblatt FJ, Klein KL; Wang MS; Lerner RL: Binding of 125I- insulin to the isolated glomeruli of rat kidney. J Clin Invest 64: 1357-1364, 1979.
- Tucker BJ, Anderson CM, Thies RS, Collins RC, Blantz RC. Glomerular hemodynamic alterations during acute hyperinsulinemia in normal and diabetic rats. Kidney Int 42: 1160-1168, 1992.
- Anderson PW, Zhang XI, Tian J, et al. Insulin and angiotensin II are additive in stimulating TGF 1 and matrix mRNA in mesangial cells. Kidney Int 50: 745-753, 1996.
- Cohen Ah. Massive obesity and the kidney: A morphologic and statistical study. Am J Pathol 81: 117-130, 1975.
- Kambham N, Markowitz GS, Valeri AM, Lin J, D’Agati VD. Obesity-related glomerulopathy: an emegin epidemic. Kidney Int 59: 1498-1509, 2001.
- May JE, Brandy MW, Henegar JR, Shek EW. Abnormal kidney function as a cause and a consequence of obesity and hypertension. Clin Exp Pharmacol Physiol 25: 58-64, 1998.
- Chagnac A,Weinstein T, Korzets A, Ramadan E, Hirsch J, Gafter U. Glomerular hemodynamics in severe obesity. Am J Physiol 278: F817-F822, 2000.
- Henegar JR, Bigler SA, Henegar LK, Tyagi SC, Hall JE. Functional and structural changes in the kidney in the early stages of obesity. JASN 12; 1211-1217, 2001.
- Bohman SO, Tyden G,Wilczek H, et al. Prevention of kidney graft diabetic nephropathy py pancreas transplantation in man. Diabetes 34: 306-308, 1985.
- Fioretto P, Mauer SM, Bilous KW,Goetz FC, Sutherland DE, Steffes MW. Effects of pancreas transplantation on glomerular structure in insulin-dependent diabetic patients with their own kidneys. Lancet 342: 1193-1196, 1993.
- Hotta O, Taguma Y, Chiba S et al. Possible relationship between hyperinsulinemia and glomerular hypertrophy in nephrosclerosis. Renal Fail 18: 271-278, 1996.
- Fliser D, Pacini G, Engelleiter R, et al. Insulin resistance and hyperinsulinemia are already present in patients with incipient renal disease. Kidney Int 53: 1343-1347, 1998
- Michel O, Heudes D, Lamarre J, et al. Reduction of insulin and triglycerides delays glomerulosclerosis in obese Zucker rats. Kidney Int 52: 1532-1542, 1997.
- Abrass CK, Peterson CV, Ragi GJ. Phenotypic expression of collagen types in mesangial matrix of diabetics and non diabetics rats. Diabetes 38: 1695-1702, 1988.
- Abrass CK, Spicer D, Raugi GJ. Induction of nodular sclerosis by insulin in rat mesangial cells in vitro. Studies of collagen. Kidney Int 47: 25-37, 1995
- Wang SN, Lapage J, Hirschberg R. Loss of tubular bone morphogenetic protein 7 in diabetic nephropathy. JASN 12: 2392-2399, 2001.
- Wang S, Chen Q, Simon TC et al. Bone morphogenic protein 7 (BMP-7), a novel therapy for diabetic nephropathy, 2003.
|