1 The Diabetes Control and Complications Trial Research Group: The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329: 977-86, 1993
2 UK Prospective Diabetes Study (UKPDS) Group: Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group. Lancet 352: 837-53, 1998
3 Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, Kojima Y, Furuyoshi N, Shichiri M.: Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract. 28:103-17, 1995
4 Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329:1456-62, 1993
5. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S; RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345:861-69, 2001
6. Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I; Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345:851-60, 2001
7 Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 47: 859-66, 1998
8 Haneda M, Araki S, Togawa M, Sugimoto T, Isono M, Kikkawa R. Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glomerular mesangial cells cultured under high glucose conditions. Diabetes 46: 847-53, 1997
9 Haneda M, Kikkawa R, Arimura T, Ebata K, Togawa M, Maeda S, Sawada T, Horide N, Shigeta Y. Glucose inhibits myo-inositol uptake and reduces myo-inositol content in cultured rat glomerular mesangial cells. Metabolism 39: 40-45, 1990
10 Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, Nyengaard JR, van den Enden M, Kilo C and Tilton RG: Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 42: 801-13, 1993
11 Brownlee M, Cerami A and Vlassara H: Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med 318: 1315-21, 1988
12 Abbott KC, Bakris GL: Treatment of the diabetic patient: focus on cardiovascular and renal risk reduction. Prog Brain Res. 139:289-98, 2002
13 Sharma K and Ziyadeh FN: Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes 44: 1139-46, 1995
14 Nishizuka Y: Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 9: 484-96, 1995
15 Craven PA, Davidson CM and DeRubertis FR: Increase in diacylglycerol mass in isolated glomeruli by glucose from de novo synthesis of glycerolipids. Diabetes 39: 667-74, 1990
16 Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W and King GL: Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A 89: 11059-63, 1992
17 Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, Bursell SE, Kern TS, Ballas LM, Heath WF, Stramm LE, Feener EP and King GL: Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 272: 728-31, 1996
18 Inoguchi T, Xia P, Kunisaki M, Higashi S, Feener EP and King GL: Insulin's effect on protein kinase C and diacylglycerol induced by diabetes and glucose in vascular tissues. Am J Physiol 267: E369-79, 1994
19 Haneda M, Kikkawa R, Koya D, Uzu T, Maeda S, Togawa M, Shigeta Y: Alteration of mesangial response to ANP and angiotensin II by glucose. Kidney Int. 44:518-26, 1993
20 Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K and King GL: Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest 100: 115-26, 1997
21 Babazono T, Kapor-Drezgic J, Dlugosz JA, Whiteside C: Altered expression and subcellular localization of diacylglycerol-sensitive protein kinase C isoforms in diabetic rat glomerular cells. Diabetes. 47:668-76, 1998
22 Kikkawa R, Haneda M, Uzu T, Koya D, Sugimoto T and Shigeta Y: Translocation of protein kinase C alpha and zeta in rat glomerular mesangial cells cultured under high glucose conditions. Diabetologia 37: 838-41, 1994
23 Fumo P, Kuncio GS, Ziyadeh FN: PKC and high glucose stimulate collagen alpha 1 (IV) transcriptional activity in a reporter mesangial cell line. Am J Physiol. 267(4 Pt 2):F632-8, 1994
24 Shankland SJ, Scholey JW: Expression of growth-related protooncogenes during diabetic renal hypertrophy. Kidney Int. 47:782-8, 1995
25 Kreisberg JI, Radnik RA, Ayo SH, Garoni J, Saikumar P: High glucose elevates c-fos and c-jun transcripts and proteins in mesangial cell cultures. Kidney Int. 46:105-12, 1994
26 Tomlinson DR: Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 42:1271-81, 1999
27 Igarashi M, Wakasaki H, Takahara N, Ishii H, Jiang ZY, Yamauchi T, Kuboki K, Meier M, Rhodes CJ and King GL: Glucose or diabetes activates p38 mitogen-activated protein kinase via different pathways. J Clin Invest 103: 185-95, 1999
28 Isono M, Cruz MC, Chen S, Hong SW, Ziyadeh FN: Extracellular signal-regulated kinase mediates stimulation of TGF-beta1 and matrix by high glucose in mesangial cells. J Am Soc Nephrol. 11:2222-30, 2000
29 Koya D, Lee IK, Ishii H, Kanoh H and King GL: Prevention of glomerular dysfunction in diabetic rats by treatment with d-alpha-tocopherol. J Am Soc Nephrol 8: 426-35, 1997
30 Isshiki K, Haneda M, Koya D, Maeda S, Sugimoto T and Kikkawa R: Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes 49: 1022-32, 2000
31 Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, Sugimoto T, Yasuda H, Kashiwagi A, Ways DK, King GL, Kikkawa R: Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 14:439-47, 2000
32 Aiello LP: The Potential Role of PKC beta in Diabetic Retinopathy and Macular Edema. Surv Ophthalmol 47 Suppl 2: S263-9, 2002
33 Vinik AI: Neuropathy: new concepts in evaluation and treatment. South Med J 95: 21-3, 2002
34 Beckman JA, Goldfine AB, Gordon MB, Garrett LA and Creager MA: Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res 90: 107-11, 2002
35 Haneda M, Koya D, Isono M, Kikkawa R: Overview of glucose signaling in mesangial cells in diabetic nephropathy. J Am Soc Nephrol. 14:1374-82, 2003

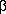 and extracellular matrix (ECM) proteins. To understand the pathophysiological significance of PKC-MAPK activation in diabetic nephropathy, the effects of PKC inhibitors, such as vitamin E (
and extracellular matrix (ECM) proteins. To understand the pathophysiological significance of PKC-MAPK activation in diabetic nephropathy, the effects of PKC inhibitors, such as vitamin E ( -tocopherol), thiazolidinediones, and a specific inhibitor for PKC
-tocopherol), thiazolidinediones, and a specific inhibitor for PKC
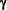 ), novel PKC (
), novel PKC (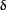 ,
,  ,
, 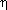 ,
, 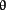 ,
, 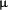 ), and atypical PKC (
), and atypical PKC (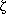 ,
, 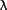 ) on the basis of their common structural features [14]. The classical PKC enzymes contain two cysteine-rich zinc finger-like motifs (C1 region), which are essential for interaction with phorbol ester and diacylglycerol (DG), and a Ca[2+]-binding domain (C2 region) in their regulatory domain. The novel PKC enzymes do not require Ca[2+] since C2 region is absent. The novel PKC enzymes are activated by phosphatidylserine and DG or phorbol esters. The atypical PKC enzymes, which lack the C2 region and one of the cysteine-rich zinc finger-like motifs in the C1 region, are not activated by Ca[2+], DG, or phorbol esters, but their activation depends on phosphatidylserine and cis-unsaturated fatty acids.
) on the basis of their common structural features [14]. The classical PKC enzymes contain two cysteine-rich zinc finger-like motifs (C1 region), which are essential for interaction with phorbol ester and diacylglycerol (DG), and a Ca[2+]-binding domain (C2 region) in their regulatory domain. The novel PKC enzymes do not require Ca[2+] since C2 region is absent. The novel PKC enzymes are activated by phosphatidylserine and DG or phorbol esters. The atypical PKC enzymes, which lack the C2 region and one of the cysteine-rich zinc finger-like motifs in the C1 region, are not activated by Ca[2+], DG, or phorbol esters, but their activation depends on phosphatidylserine and cis-unsaturated fatty acids.