
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
Congenital Obstructive Nephropathy: Lessons from animal models
Helen Liapis MD and Sanjay Jain MD, PhD
Department of Pathology and Immunology,
Washington University, St. Louis. MO, USA
Congenital obstructive nephropathy is a common disease in infants and children affecting approximately 1% of all newborn. Obstruction occurs in utero and disease severity depends on the time of onset. Early onset obstruction leads to partial or complete renal dysplasia, defined as disorganization of the kidney architecture with failure to differentiate into glomeruli and tubules. Obstruction later in gestation is generally accompanied by milder kidney damage. Obstruction of urine flow may be at the level of uretopelvic junction (UPJ), at any length of the ureter, or in the bladder and or the urethra. Obstruction of urine flow invariably damages the kidney parenchyma. Bilateral obstruction is the most severe form and progresses to chronic renal failure early in childhood. Obstructive nephropathy is a leading cause of primary renal disease in children on dialysis or with a kidney transplant1. Surgically resected human kidneys with congenital obstructive nephropathy show a spectrum of pathologic features, such as hydronephrosis and complete or partial multicystic dysplasia (Figure 1) 2.  UPJ obstruction may be associated with glomerulosclerosis, variable tubulointerstitial fibrosis or milder histopathologic features in low grade obstruction3. Most of the obstructive anomalies are unilateral and occur predominantly in males in the newborn period. However, there is a female preponderance later in childhood 4. A familial occurrence is rare, but well recognized5. The molecular and genetic basis and the critical events in kidney development that are disrupted by obstruction are poorly understood. Furthermore, obstruction in some cases (for example in familial disease) may not be the cause but the result of inherent germ line or somatic mutations, opening up an interesting line of investigation. Elucidating these mechanisms is crucial to our understanding of obstructive nephropathy and it will be essential for development of cures, as the current therapeutic approaches have had only limited success. Animal models have been extremely valuable in contributing to our understanding of the pathophysiology of obstruction. There are three major experimental animal model categories 6 :
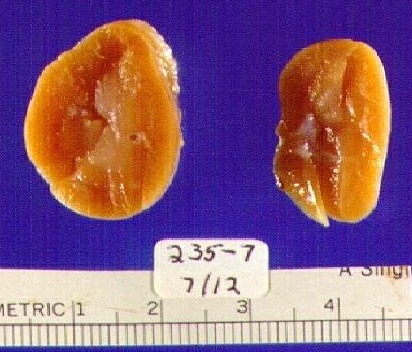
Surgical models for fetal urinary obstruction Models of surgical ligation of one ureter are often referred to as UUO (for unilateral ureteral obstruction). A major advantage of UUO is its reproducibility. These models have been around for decades before genetically engineered animals became widely available, and have proven important for the study of altered cellular and molecular phenomena in the obstructed kidney. They continue to be useful for the evaluation of potential therapeutic agents to ameliorate renal damage. Neonatal rat, rabbit, opossum, fetal ship and fetal pig have been successfully obstructed to recapitulate the disease in humans6. The kidney in most of these animals at the time of birth is at the mesonephric stage of development and it roughly corresponds to second trimester human equivalent. In the neonatal rat and rabbit the major feature of UUO is hydronephrosis 7 (Figure 2). 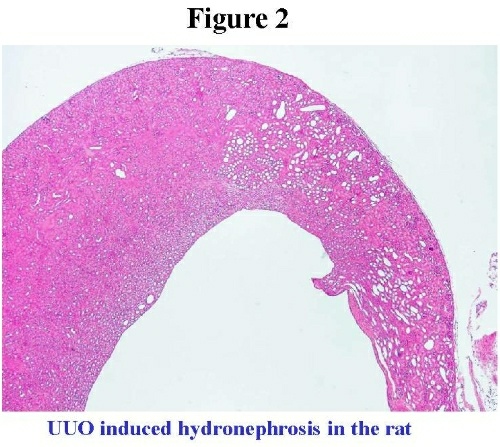 The fetal opossum and Molecular Pathophysiology of congenital obstruction We have used the North American opossum pup, a marsupial (Figure 3). The pups have mesonephric kidneys at birth, 12.5 days post conception. Complete unilateral ureteral obstruction is performed at 20 days of age when pups are mature enough to sustain surgery. Animals are sacrificed at two weeks post-obstruction.  Obstructed (Ob) kidneys are typically smaller compared to unobstructed contralateral kidney (Figure 4). Figure 4
 Obstructed kidneys show cystic tubules and expansion of the interstitial mesenchymal cells reminiscent of renal dysplasia in humans (H+E stained sections in Figure 5). 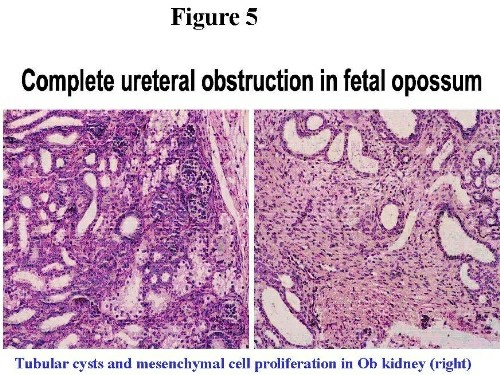 In the two week interval, there is pronounced increase in Platelet derived growth factor (PDGFA), collagen I and Fibronectin 8,9. PDGFA is localized within dysplastic epithelia and interstitial fibroblasts and co-localizes with Collagen I suggesting its role in interstitial fibrosis in obstructed immature opossum kidney. In the long term obstructed model (up to 8 weeks) Fibronectin is progressively upregulated in the interstitium in contrast to sham kidney in which Fibronectin decreases with maturation. A-sma and Collagen IV also increases progressively in Ob kidneys, while Laminin shows a constant increase (Figure 6) 10,11. 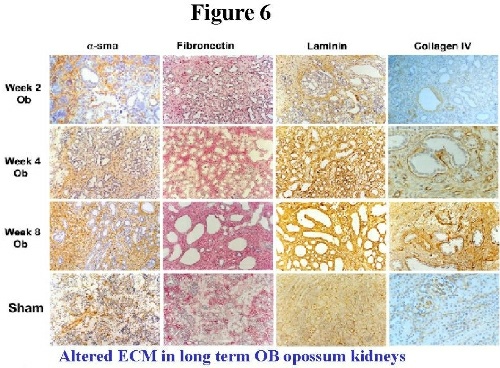 We have proposed that sustained production of Fibronectin may significantly alter the scaffold on which branching of the ureteric bud takes place. Additionally, this abnormal scaffold may enhance epithelial apoptosis, therefore linking diminished kidney growth and maturation to apoptosis in this model. Support from this comes from the observation that anti-apoptotic gene Bcl2 is decreased and the pro-apoptotic gene Bax is upregulated in these kidneys (Figure 7, left and right upper panel respectively, arrows indicate condensed apoptotic nuclei). Propidium iodide highlights apoptosis9. Chevalier etal, have found similar changes in the rabbit indicating that dysregulated apoptosis is a major event in congenital obstructive nephropathy12. 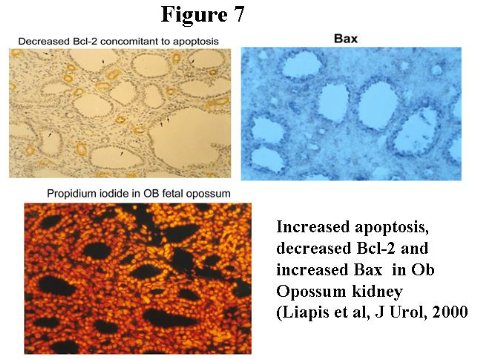 Other growth factor changes include TGF
A number of genetically engineered mutant mice have kidneys histologically similar to humans. For simplicity in this discussion, we have separated kidney development into two stages, inductive and post-inductive, and provide representative examples of genes that are important in these processes. These include: Lim-1, Eya1, WT1, Pax-2, cRET, Wnt-4, PDGFB, PDGFRB, Angiotensin type 2 receptor (AT2) and GDNF. In induction, WT-1, PAX-2, GDNF and cRET are important (Table below). Interestingly, in some of these mice, gene dosage may play an important role, as heterozygous mice (for example, GDNF) also show a subset of features of kidney defects seen in the homozygous mice. Regarding the post-induction genes, Wnt-4 is normally expressed in the mesonephric mesenchyme surrounding the primitive ducts. Later in development Wnt-4 localizes at sites of new tubule formation. Mutant Wnt-4 mice are small in size and have hypoplastic kidneys with no glomeruli, suggesting that the mesenchyme is induced but, it is not transformed into epithelia. Hence, these findings support a role for Wnt-4 in the generation of glomeruli and tubules13, 14. Bone morphogenitic protein -7 (BMP-7), a member of the TGF AT2 receptor is an embryonic gene of particular interest. It is expressed by mesenchymal cells in the early mesonephros. Upregulation of its transcription activity coincides with increased apoptosis of excess mesenchymal cells, a physiologic phenomenon in normal kidney development. It is proposed that failure of AT2 to enhance apoptosis will hinder the very first interactions between the ureteric bud and the condensing mesenchyme or lead to ectopic ureteral orifice (a cause of ureteral obstruction and subsequent vesicoureteral reflux or dysplasia)17,18. The table below depicts a partial list of mutant mice with disturbed kidney development. Representative genes important for kidney development
Animals with spontaneous mutations and congenital hydronephrosis are relatively few. Unilateral hydronephrosis occurs spontaneously in the rat and mimics UPJ obstruction, but the pathology in this animal is poorly described 6. However, presence of spontaneous hydronephrosis in this model raises the question of whether it is possible to have hydronephrosis without urine flow obstruction. A mouse with spontaneous severe, unilateral UPJ is also described however the studies were hindered due to difficulties in breeding 19. Congenital obstruction in humans Obstructive nephropathy in humans is a heterogeneous disease with a histopathogic spectrum. There are relatively few studies that have utilized human tissue to identify genes that are abnormally regulated in the affected kidneys. Some of the genes we studied are in general agreement with findings in animals 20,21. Our material was derived from surgically resected multicystic kidneys that had characteristic histology of renal dysplasia. The classic features of dysplasia are randomly arranged cysts, both cortical and medullary, concentric fibromuscular collars around primitive ducts, architectural disorganization of the entire kidney, chronic inflammatory infiltrates, severe decrease in functioning nephrons, and in about 30% of cases metaplastic cartilage Some of these features, for example hydronephrosis, interstitial inflammation and cysts are replicated in some animal models. However, in no animal model metaplastic cartilage has been reproduced. Fibromuscular collarettes are another feature that most experimental models do not have (Figure 1, arrows). However, development of the mammalian kidney is essentially conserved and gene defects in mutant mice or altered gene expression in surgical UUO models are likely to be shared by humans. In our GeneChip analysis of human dysplatic kidneys, we compared dysplastic and normal age matched human kidneys and found significant differences in gene expression. An example of genes altered by >2fold is shown in a heat map in Figure 8. Relevant altered genes fall into four general categories: 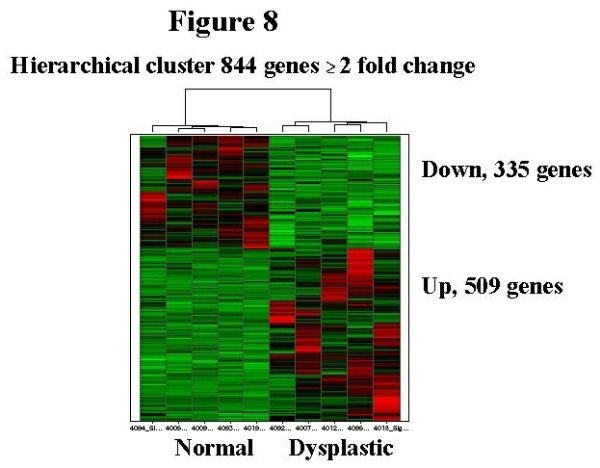
A sample of structural, extracellular matrix genes and growth factors is diagrammatically shown in Figure 9. Renin, WT1, Angiotensin Receptor 2, Glypican 3, Sall1, BMP7, PDGFA, and Tyrosine Kinase (TYR03), Collagen II, MMP7 2. Among these genes many were previously studied in animals and important physiologic functions were revealed. For example, Glypican -3-deficient mice have cystic dysplastic kidneys and verall increased body growth similar to Beckwith-Wiedemann syndrome, known to be frequently associated with urogenital malformations including renal dysplasia. 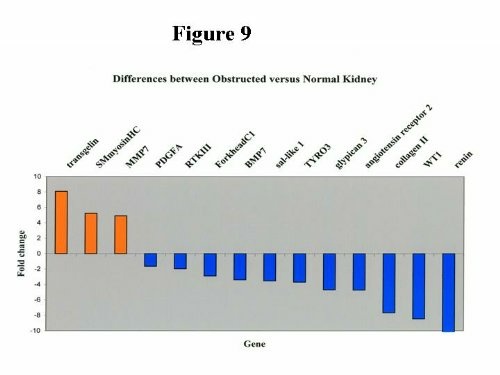 Defects in BPM7, AT2R and Sall 1 deficient mice were described above. Little is known on the role of RTKIII, forkhead C of the winged helix family of transcription factors in kidney development. These and other genes may be potentially significant in humans. Gene expression profiles provide an opportunity to describe dysplasia in molecular terms. Recently, the first report of GeneChip arrays in obstructed rats was published. Multiple immune regulators and structural genes were found altered, some detected for the first time22. Conclusions In conclusion, animal studies have provided unprecedented insights in the pathogenesis of congenital obstructive nephropathy. New technologies such as GeneChip arrays are revolutionizing our ability to examine human tissue and generate an overwhelming amount of data that need to be carefully and methodically analyzed. A general agreement in altered gene expression in humans is suggested by the findings in animal models. The next step will be to define the genetic regulatory cascade of genes altered by obstruction and to understand the intriguing links among the many, seemingly redundant signaling systems and how they perform in concert, in vivo. This area of research is of paramount importance in identifying new disease modifiers with potential therapeutic implications. Questions being addressed in models of congenital obstructive nephropathy that pertain to humans include:
1. Warady, B.A., et al., Renal transplantation, chronic dialysis, and chronic renal insufficiency in children and adolescents. The 1995 Annual Report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Nephrol 1997, 11(1): p. 49-64. 2. Liapis, H. Biology of congenital obstructive nephropathy. Nephron Exp Nephrol, 2003. 93(3): p. e87-91. 3. Zhang, P.L., C.A. Peters, and S. Rosen. Ureteropelvic junction obstruction: morphological and clinical studies. Pediatr Nephrol 2000, 14(8-9): p. 820-6. 4. Pope, J.C. 4th, Brock JW 3rd, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol 1999, 10(9): p. 2018-28. 5. Roodhooft, A.M., J.C. Birnholz, and L.B. Holmes. Familial nature of congenital absence and severe dysgenesis of both kidneys. N Engl J Med 1984, 310(21): p. 1341-5. 6. Peters, C.A. Animal models of fetal renal disease. Prenat Diagn 2001, 21(11): p. 917-23. 7. Cruz RC, Steinhardt GF, Liapis H, et al. The role of matrix metalloproteinases (MMPs) in unilateral ureteral obstruction (UUO) in newborn rats. FASEB J 16 (5): A1097 Part 2, 2002. 8. Liapis, H., M. Nag, and G. Steinhardt. Effects of experimental ureteral obstruction on platelet-derived growth factor-A and type I pro-collagen expression in fetal metanephric kidneys. Pediatr Nephrol 1994, 8(5): p. 548-54. 9. Liapis, H., H. Yu, and G.F. Steinhardt.Cell proliferation, apoptosis, Bcl-2 and Bax expression in obstructed opossum early metanephroi. J Urol 2000, 164(2): p. 511-7. 10. Drakopoulos A Charonis A, Vlachojiannis J, Liapis H. Differential expression of endothelin receptors and increased extracellular matrix synthesis in the developing kidney following unilateral ureteral obstruction in the opossum. Nephrol Dial Transpl Vol 18 Supplement, p 281(M878A), 2003 11. Liapis H, Barent B, Steinhardt GF. Extracellular matrix in fetal kidney after experimental obstruction. J Urol. 2001, 166(4):1433-8. 12. Chevalier, R.L. Molecular and cellular pathophysiology of obstructive nephropathy. Pediatr Nephrol 1999, 13(7): p. 612-9. 13. Piscione TD, Rosenblum ND. The molecular control of renal brunching morphogenesis: Current knowledge and emerging insights. Differentiation 2002, (7):227-246. 14. Woolf AS, Winyard PJ: Moleculae mechanisms of human embryogenesis: Developmental pathogenesis of renal tract malformations. Pedeatr Dev Pathol 2002, (5)108-129. 15. Jena N, Martin-Seisdedos C, McCue P, Croce CM. BMP7 null mutation in mice: developmental defects in skeleton, kidney, and eye. Exp Cell Res. 1997, J 10; 230(1):28-37. 16. Kiefer SM, Ohlemiller KK, Yang J, McDill BW, Kohlhase J, Rauchman M. Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum Mol Genet. 2003, 1;12(17):2221-7. 17 Matsusaka T, Ichikawa I. Biological functions of angiotensin and its receptors. Annu Rev Physiol. 1997, 59:395-412. 18 Lipschutz JH. Molecular development of the kidney: a review of the results of gene disruption studies. Am J Kidney Dis 1998, 75(1)30:-36. 19 Horton Jr CE, Davisson MT, Jacobs JB, Bernstein CT, Retic AB, Mandell J. Congenital progressive hydronephrosis in mice: a new recessive mutation. J Urol 1988, 140:1310-1305. 20. Liapis, H., Doshi, R.H., Watson, M.A., Liapis, A., Steinhardt, G.F. Reduced renin expression and altered gene transcript profiles in multicystic dysplastic kidneys. J Urol 2002, 168(4 Pt 2):1816-20. 21. Suarez A, Steinhardt GF, Liapis H: Altered gene transcript profiles in congenitally obstructed human kidneys. Lab Invest 2003, (83), A1227. 22. Silverstein DM, Travis BR, Thornhill BA, Schurr JS, Kolls JK, Leung JC, Chevalier RL. Altered expression of immune modulator and structural genes in neonatal unilateral ureteral obstruction. Kidney Int. 2003, 64:25-35. |
 which is increased, IGF and Renin/Angiotensin, which are consistently decreased in various models12. Our studies and studies by others suggest that given the seemingly functional redundancy of many factors acting in concert in the kidney, the subtotal of altered factors may be provide more useful information than individual factors per se. New technologies, such as CeneChip arrays present an unprecedented opportunity to examine multiple genes simultaneously utilizing mathematical data analysis (see below under obstruction in humans).
which is increased, IGF and Renin/Angiotensin, which are consistently decreased in various models12. Our studies and studies by others suggest that given the seemingly functional redundancy of many factors acting in concert in the kidney, the subtotal of altered factors may be provide more useful information than individual factors per se. New technologies, such as CeneChip arrays present an unprecedented opportunity to examine multiple genes simultaneously utilizing mathematical data analysis (see below under obstruction in humans). 3
3