
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
BASES MOLECULARES DE LA BARRERA DE FILTRACION GLOMERULAR-SINDROME NEFROTICO CORTICORRESISTENTE.
STELLA MARIS DIEGUEZ
El glomérulo es responsable de la producción de la orina a partir de la elaboración del ultrafiltrado plasmático. La capacidad de filtración de la barrera glomerular tiene una doble naturaleza: mecánica y eléctrica
Una pequeńa cantidad de proteínas se encuentra presente normalmente en la orina, en su la mayoría derivan del plasma, otras se originan en el tejido renal. La composición final de las proteínas en la orina, tanto en sujetos sanos como en las enfermos, es el resultado neto de tres funciones: filtración glomerular, reabsorción tubular y la adición o secreción de proteínas a la orina a través del tracto genitourinario. La perturbación de estos mecanismos conduce a la presencia de proteinuria. Para evaluar si ésta se debe a una alteración de la filtración es necesario conocer la estructura del capilar glomerular.
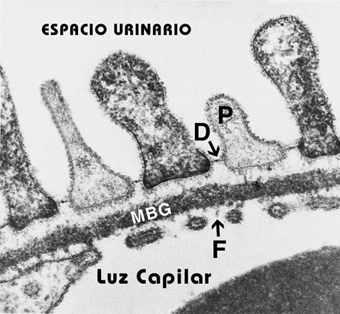
La pared capilar glomerular es una barrera molecular capaz de excluir a la mayoría de las proteínas plasmáticas y permitir el paso del agua, de pequeńas moléculas de soluto y de iones. Entre la sangre y el espacio urinario, una sustancia, debe atravesar la barrera de filtración glomerular compuesta por: el endotelio fenestrado, la membrana basal glomerular y la hendidura del poro y la zona que queda entre los procesos pedicelares de los podocitos1 En la fotografía siguiente (Figura 1), se puede observar la ubicación de las tres fases de la filtración: el endotelio con fenestraciones (F), membrana basal glomerular (MBG) y epitelio visceral, formado por los podocitos (P) que dejan ver el diafragma (D) entre los pedicelos. Figura 1
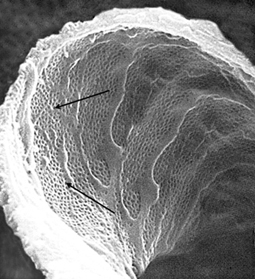
 EL ENDOTELIO El endotelio está perforado por poros o fenestraciones que permiten la separación mecánica de los elementos de la sangre y el plasma. Los poros miden 70 y 100 nm de diámetro como se ve abajo en la fotografía (Figura 2), donde las flechas indican los numerosos poros endoteliales. Figura 2
 La superficie de la célula endotelial está cargada negativamente por la presencia de una glucoproteína polianiónica, la podocalixina, que es la principal sialo-proteína glomerular. La aglomeración de moléculas superficiales aniónicas y fenestraciones hace que el endotelio glomerular se diferencie de otras membranas plasmáticas endoteliales y que permita el paso de moléculas de bajo peso molecular2. Aunque no es muy eficiente para impedir el pasaje de macromoléculas.
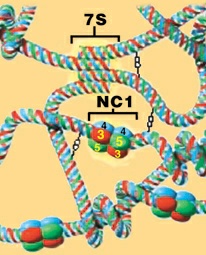
La membrana basal glomerular (MBG), impide el paso de macromoléculas en forma mecánica y eléctrica; esta última por la presencia de carga es negativa, proteoglicanos ricos en heparán sulfato. Los estudios con dextranos han sugerido que la integridad estructural de la MBG es clave para el mantenimiento de la función de permeabilidad de la barrera al agua, pequeńos solutos, iones, y proteínas de menor tamańo. Sin embargo no lo es para proteínas plasmáticas mayores de 70 kDa (3. La MBG se compone de dos capas finas, la lámina rara interna y la lámina rara externa, y una capa central gruesa, la lámina densa. Las células endoteliales y epiteliales adyacentes secretan moléculas tales como colágeno tipo IV, laminina, fibronectina, nidogén/enactina, y proteoglicanos de heparán sulfato que forman una estructura, semejante a un enrejado. Hay sitios aniónicos, los glucosaminoglicanos de heparán sulfato, en las tres capas que componen la MBG. Si estos se remueven, se incrementa la permeabilidad de la membrana basal glomerular. El colágeno tipo IV es el mayor constituyente colagenoso de la membrana basal MBG.4. Se trata de un heterotrímero que consta de un dominio carboxiterminal no colagenoso (NC1). Las moléculas del colágeno IV pueden asociarse a través de este dominio para formar dímeros, y por medio de sus terminaciones amino formar tetrámeros.4;37 Las macromoléculas de colágeno tipo IV compuesto predominantemente por las cadenas con isoformas Figura 3
 Las hebras que forman la red consisten en agregados de, al menos, cinco sustancias: el colágeno tipo IV, tres glucoproteínas: laminina, nidogén y fibronectina, y un proteoglicano el heparán sulfato.6 Figura 4
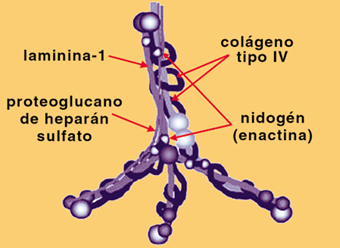 En el esquema anterior (Figura 4) de Mohamed E et al.6 se observan las cadenas del colágeno conectadas por el dominio S7. En el se entrelazan las moléculas de laminina, nidogén y fibronectina, y un proteoglicano el heparán sulfato Los brazos están abiertos simétricamente en el espacio. En la figura 5, abajo, observamos, la red tridimensional que compone la MBG es una estructura poligonal con poros que tienen de 4 a 6 nm de diámetro. Figura 5
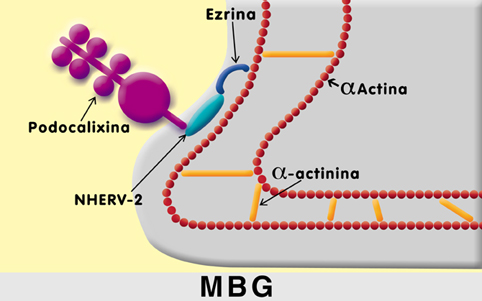
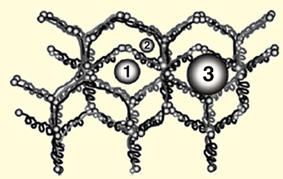 Esquema descripto por Mohamed E et al6 EL PODOCITO Y EL PORO DE FILTRACIÓN GLOMERULAR El tercer elemento de la barrera de filtración glomerular lo constituyen las células epiteliales viscerales o podocitos, encargados de sintetizar la MBG y formar los poros de filtración. Los podocitos son células muy diferenciadas que no se dividen. Existiría un número de podocitos inicial, que se pierden de forma progresiva e irreversible en el transcurso de una lesión glomerular8. Aunque, en algunas glomerulopatías como las colapsantes, el fenotipo del podocito se altera y es capaz de dividirse9. Las células epiteliales expresan una serie de proteínas específicas que son indispensables para el mantenimiento de la compleja estructura de la barrera de filtración, de los procesos pedicelares interdigitados y del diafragma de hendidura (pequeńas hendiduras cubiertas por una película proteica).10 La superficie del podocito podría ser dividida en tres dominios con diferentes localizaciones, componentes proteicos y funciones10. En cada dominio existen proteínas, que son fundamentales para el mantenimiento y la integridad del mismo, más aún, para la estabilidad global de la arquitectura del podocito.11 DOMINIO DE SUPERFICIE DEL PODOCITO –SUS PROTEINAS Dominio apical: podocalixina, ezrina, complejo NHERF-2 (cubren la superficie del podocito). Dominio del diafragma de filtración: la principal responsable de la propiedad de selectividad del diafragmaes la nefrina. A este nivel también encontramos a P-cadherina, neph-1, podocina, CD2AP, ZO-1, filtrina, etc. Dominio basal o de anclaje es el encargado de fijar al pedicelo a la membrana basal glomerular . Encontramos el complejo distroglicano, el complejo integrina DOMINIO APICAL La superficie de los podocitos está cubierta por carga eléctrica negativa, siendo la podocalixina la mayor de las sialoproteínas de los mismos. Esta es una proteína de membrana, polianiónica, importante en el establecimiento de la carga negativa glomerular, en el mantenimiento de la arquitectura celular y de la distancia intercelular4. La podocalixina esta disminuída en la glomerulonefritis focal y segmentaria (GNSF) y normal en el síndrome nefrótico a cambios mínimos (SNCM). Por medio de la microscopía inmunoelectrónica se vio que la podocalixina contacta con la ezrina, una proteína intracelular, miembro de la familia ERM, proteínas ligadoras de actina.12. Esto sugiere que la podocalixina puede estar asociada con la extensa red de filamentos de actina y de esta forma participa en el mantenimiento de la estructura podocitaria y del espacio intercelular. La podocalixina está unida a la ezrina y a la actina del citoesqueleto a través de un factor 2 regulador del intercambiador Na+ / H+, la NHERF213 Si las interacciones podocalixina / NHERF2 / zezrina / actina se rompen aparecen cambios en los procesos pedicelares de la célula epitelial glomerular y ésto se asocia a patología glomerular.13 En la figura 6, modificación del esquema de Kerjaschki D. y col.11, se puede observar la ubicación de la proteínas y su conexión con la actina, proteína que mantiene la estructura podocitaria . Figura 6
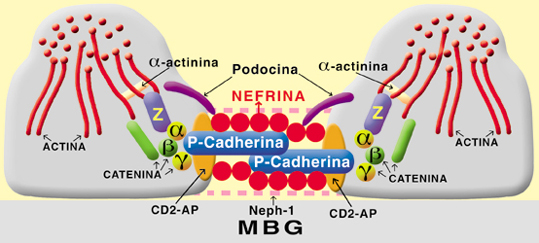
 DOMINIO DEL DIAFRAGMA DE FILTRACIÓN Entre los procesos pedicelares que cubren la superficie externa de la membrana basal glomerular existen hendiduras de 25 a 60 nm que están cruzadas por una membrana delgada llamada diafragma de hendidura o diafragma de filtración. Esta fina estructura es la responsable principal de impedir el paso de moléculas como la albúmina.4 Los poros son rectangulares, de aproximadamente. 40 Ä por 140 Ä en la sección transversal, y 70 Ä en la sección longitudinal.1 El diafragma de filtración exhibe una subestructura similar a la de un "cierre relámpago". Este modelo fue propuesto por Rodewald y Karnowsky4, constaría de puentes alternantes que se extienden desde la membrana plasmática de un podocito a otro. Tendría un filamento central que corre paralelamente y en forma equidistante a las membranas celulares. El mayor componente del diafragma de filtración es la nefrina producto de un gen llamado NPHS1. Esta glucoproteína transmembrana tipo 1 pertenece a la superfamilia de las inmunoglobulinas (Ig) y tiene ocho módulos similares a los de las Ig y un módulo simple de fibronectina tipo III.4 Si se inhibe su acción mediante los anticuerpos antinefrina, la estructura del diafragma de filtración persiste, pero se altera la filtración4; 14. Estos hallazgos indican que la nefrina no es esencial para el mantenimiento de la morfología ultraestructural del diafragma de filtración, pero sí lo es para sostener su función. La nefrina podría interactuar con el centro proteico del diafragma de filtración, fundamentalmente con la P-cadherina4. La P-cadherina tiene un dominio extracelular que forma esencialmente el andamiaje del diafragma de filtración aunque no es la única que sirve como estructura básica del mismo. El dominio intracelular está conectado con la En la periferia de la membrana sobre la superficie citoplasmática de los diafragmas de filtración en los sitios de unión se encuentra la proteína ZO-1, que pertenece a una familia llamada "guanilato-kinasa asociadas a membranas" y es una variante de las uniones intercelulares. Existe una nueva subfamilia de moléculas "nefrina like" (parecidas a la nefrina) que incluye a la neph 1, está localizada exclusivamente en las márgenes laterales de los procesos pedicelares de los podocitos, en el lugar de inserción del diafragma de filtración.16. La neph 1 y la nefrina podrían interactuar a través de las uniones intercelulares de los procesos pedicelares. La podocina es otra molécula que está localizada en la fase citoplasmática del diafragma de filtración.17; 18 junto con la CD2AP, proteína asociada al CD2 primeramente descripta en los linfocitos T. A nivel del dominio del diafragma de filtración fijan la nefrina al citoesqueleto de actina11. El complejo de esta unidad funcional del diafragma de filtración y su ensamble al podocito estaría formado por nefrina-podocina-CD2AP. Un nuevo gen llamado NLG1, la filtrina, podría ser una molécula que está fuertemente asociada a la modulación de las propiedades de la nefrina.19 En la figura 7 vemos la interacción de las proteínas que componen el diafragma de filtración en lazadas como un cierre relámpago y su conexión con la Figura 7
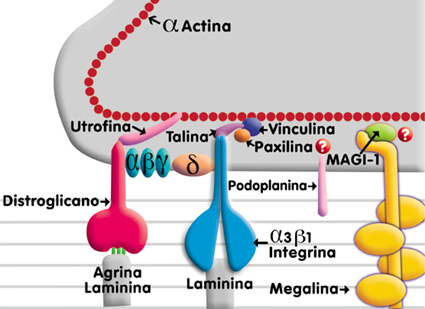
La membrana basal del podocito contiene algunas proteínas de adhesión que une a los podocitos con la matriz extracelular11 El complejo de adhesión esta formado por complejo El complejo distroglicano posee dos subunidades: La proteína MAGI-1 se asocia con la megalina, la cual es un receptor poliespecífico multiligando, cuya función es de receptor endocítico para lipoproteínas. La asociación intracelular de la megalina con el MAGI-1 podría ser un complejo de unión adicional.11 En la figura 6 vemos en un esquema modificado del propuesto por Kerjaschki D.11 los componentes del complejo de anclaje. Figura 8
 DISFUNCIÓN DE LA BARRERA DE FILTRACIÓN: ALTERACIONES MOLECULARES Y PROTEINURIA El desarrollo de proteinuria es la manifestación cardinal de la injuria glomerular y un factor patogénico de progresión de la disfunción renal. El incremento de la excreción urinaria de proteínas es el resultado de alteraciones en la permeabilidad selectiva de la pared del capilar glomerular o de su manejo tubular. La mayoría de los estudios experimentales realizados muestran que la alteración de la selectividad de la pared capilar glomerular es una combinación de pérdida de carga eléctrica negativa y de aumento del tamańo del poro.20 La restricción al paso de proteínas se basa en el tamańo, la carga eléctrica y la configuración estérica de la partícula.20. En condiciones fisiológicas, las proteínas de tamańo similar al de las inmunoglobulinas (IgG), con cargas neutras y radio molecular de 55 Ä, no se filtran debido a que sus tamańos son más grandes que los de los poros de filtración. La albúmina, un anión, cuyo radio molecular es de 36 Ä tiene baja permeabilidad, su restricción a la filtración es debido a que es repelida por la carga eléctrica negativa de la pared del capilar glomerular. Dicha carga está dada por la podocalixina, del endotelio y del podocito, y por el heparán sulfato ubicado en el ámbito de la membrana basal glomerular.20 ALTERACIONES MOLECULARES DE LA MEMBRANA BASAL GLOMERULAR La mutación de la cadena En el cuadro siguiente se describen las alteraciones genéticas y síndromes clínicos descriptos en los que hay alteración de las cadenas del Colágeno IV37
ALTERACIONES MOLECULARES DEL DIAFRAGMA DE FILTRACION NEFRINA Y DEL NEPH-1 Se han descripto más de 30 alteraciones del gen de la nefrina. En animales de experimentación la falta de nefrina muestra síndrome nefrótico neonatal. Ratones homocigotas con el gen de Neph1 inactivado nacieron semejantes a los normales y inmediatamente desarrollaron proteinuria masiva con edema y murieron en 24 horas. Los rińones tenían agrandamiento del espacio de Bowman, túbulos dilatados, borramiento de los pies de los podocitos, ausencia del diafragma podocitario. Los mismos hallazgos se encuentra en pacientes con alteración del gen NPHS1 humano. El único defecto encontrado es la pérdida de nefrina en la célula podocitaria Gen NPHS1 cromosoma 19 q13.1 (NEFRINA) SÍNDROME NEFRÓTICO CONGÉNITO DE TIPO FINLANDÉS (SNF): Es una enfermedad caracterizada por proteinuria masiva en el periodo perinatal. Los paciente presentan la eliminación completa de los procesos pedicelares en la microscopía electrónica. La inmunofluorescencia y la microscopía inmunoelectrónica demostraron que existe una pérdida casi completa de expresión de la nefrina en esta enfermedad congénita8, 32. En el SNF la ausencia de función de la nefrina es crucial para el desarrollo de proteinuria masiva, incluso en etapas tan tempranas como la intrauterina. Los pacientes presentan síndrome nefrótico corticoresistente desde el comienzo de la vida y evolucionan a la insuficiencia renal terminal . El único tratamiento es el transplante. En el SNF la síntesis de nefrina está disminuida, y además existe una redistribución a nivel ultraestructural.23 NEFRINA EN LAS ENFERMEDADES GLOMERULARES ADQUIRIDAS Actualmente se plantea que la nefrina juega un papel importante en la patogénesis de algunas enfermedades glomerulares. Por otro lado la expresión de la nefrina podría cambiar o desaparecer con la alteración estructural de los podocitos. Los mecanismos y las consecuencias de la lesión del podocito son todavía mal conocidos. Uno de los aspectos patogénicos más importantes de las nefropatías es la lesión y pérdida de podocitos, con la consecuente "insuficiencia" podocitaria y colapso del capilar glomerular.24 Hay dos tipos de lesión de esta célula con consecuencias clínicas: la lesión subletal de las nefropatías potencialmente reversibles (cambios mínimos y en la nefropatía membranosa) y la lesión letal que daría lugar a una nefropatía progresiva.8; 25 Lesión letal se observa en la glomeruloesclerosis focal y segmentaria donde existe una pérdida urinaria de podocitos que permite distinguirla del síndrome nefrótico de cambios mínimos8. En las glomerulopatías colapsantes como la nefropatía por HIV los podocitos pierden marcadores de diferenciación, dejan de comportarse como tales y aumenta su tasa de proliferación y apoptosis.9 La pérdida de podocitos por apoptosis es la manera más frecuente de pérdida de células en fisiopatología renal26. Las células apoptóticas pierden el contacto con células adyacentes y con la matriz extracelular, y la morfología apoptótica se mantiene durante un período de tiempo muy corto (1-2 horas). Dado que están expuestos a una presión de 40 mmHg son arrastrados por el flujo urinario. A pesar de estas dificultades, estudios recientes han identificado la apoptosis de podocitos como un hecho característico en modelos animales del síndrome nefrótico, como el producido por la puromicina23; 28. FACTORES QUE INCREMENTAN LA APOPTOSIS La angiotensina II tienen receptores en los podocitos e induce apoptosis. La hiperglucemia modifica la expresión de proteínas reguladoras de la apoptosis e induce apoptosis en células tubulares renales, células endoteliales y en el blastocisto. El factor transformante de crecimiento Los radicales libres de oxígeno son también mediadores de la apoptosis, se producen durante la lesión glomerular e inducen la aparición de proteinuria.8. LA LESIÓN DE PODOCITOS EN NEFROPATÍAS PROTEINÚRICAS QUE PUEDEN ASOCIARSE A UNA DISMINUCION DE LA EXPRESION DE LA NEFRINA NEFROPATIA DIABETICA La expresión de nefrina está reducida en los glomérulos de pacientes con nefropatía diabética. Los inhibidores de la enzima convertidora de la angiotensina II (IECA) incrementan de la expresión de la nefrina y reducen la excreción urinaria de proteínas.29 El efecto antiproteinúrico de los IECA y de los bloqueantes de los receptores AT1 de la angiotensina es considerado un factor muy importante de protección renal en pacientes con nefropatía diabética y no diabética. Recientemente se han encontrado receptores para angiotensina II en el podocito tanto en cultivo como en glomérulos intactos29. Langham y colaboradores29 han realizado un trabajo donde demuestran que la inhibición de la enzima convertidora atenúa la proteinuria a través de la reducción del radio de los poros de membrana. Aunque esos resultados obtenidos sugieren que la modulación de la expresión de la nefrina es un determinante importante de la proteinuria, otros mecanismos también contribuyen, como por ejemplo, los cambios en la composición de la membrana basal glomerular, los factores hemodinámicos, y hasta las funciones de absorción del túbulo proximal.29 SINDROME NEFROTICO A CAMBIOS MINIMOS: Esta enfermedad se presenta con frecuencia en los nińos en edades tempranas, se caracteriza por no presentar alteraciones en la microscopía óptica ni en la inmunofluorescencia. En la microscopía electrónica se ve la característica alteración de los pies de los podocitos. En la mayor parte de los casos la proteinuria remite con el uso de corticoides Se ha observado varios grados de eliminación de los procesos pedicelares, no solo entre los distintos pacientes, sino entre los glomérulos de un mismo paciente, y más aún entre los distintos segmentos de un mismo glomérulo. Estos cambios segmentarios están exacerbados por el uso de tratamiento esteroideo. Por ello cuando se analiza el papel de la nefrina en la patogénesis de la proteinuria en el SNCM su expresión debe correlacionarse con el estado ultraestructural de la barrera de filtración, por ejemplo, la extensión de los podocitos cuyos pedicelos están eliminados.29 En la mayoría de las nefropatías proteinúricas del adulto, la pérdida de pedicelos es considerada una manifestación temprana de un espectro continuo de lesión del podocito que incluye vacuolización, formación de pseudoquistes, desprendimiento del podocito de la membrana basal, y, finalmente, pérdida del podocito.30 La excepción a este hecho sería el síndrome nefrótico de cambios mínimos, donde no se ha apreciado pérdida de podocitos ni evolutividad de la nefropatía30. FILTRINA NLG1 19q13.1 El mapeo y análisis del genoma encontró el gen NLG1 en el cromosoma 19q13.1, en la adyacencia del gen NPHS1 pero que transcribe en dirección opuesta. Aún no hay alteraciones específicamente asociadas a su alteración. Como se ya se mencionó regularía la función de la nefrina. PODOCINA –GEN NPHS2-1q25-q3133 EL SÍNDROME NEFRÓTICO CORTICORRESISTENTE (SNCR) El gen mutado se llama NPHS2, está ubicado en el cromosoma 1q25-q31. La expresión de este gen está restringida a los podocitos, y el producto del mismo es la podocina.23 Boute y col. clonaron en la región 1q25-q31 el gen causante de síndrome nefrótico esteroideo-resistente Este grupo identificó 10 diferentes mutaciones del gen NPHS2 en 14 familias con SNCR. Esta enfermedad que se observa en nińos, tiene herencia autosómica recesiva, la proteinuria es resistente a los corticoides y la evolución a la insuficiencia renal terminal es rápida.23 En la mayoría de los casos la histopatología muestra glomerulonefritis con esclerosis focal y segmentaria (GNSF). Esta enfermedad no se repite en el transplante. Boute y col. detectaron mutación en la codificación del NPHS1 en pacientes con SNF y mutaciones NPHS2 en otros pacientes con el mismo síndrome. Los autores proponen un espectro de mutación NPHS1/NPHS2 que caracteriza una herencia digénica de estos genes, resultando en una modificación del fenotipo desde el SN finlandés a la GNFS congénita.
EL SÍNDROME NEFRÓTICO CORTICORRESISTENTE En individuos de familias afectadas con glomeruloesclerosis focal y segmentaria encontraron que mutación de un nucleótido en el cromosoma 19q que causaba alteración del ARN ACTN4 . En la glomeruloesclerosis focal y segmentaria autosómica dominante tipo 1 el gen mutado es el 19q 13 codifica una proteína intracelular llamada La Las mutaciones dominantes del gen podrían conducir a una mayor afinidad por los filamentos de actina y, por lo tanto, interferir con el normal ensamble / desensamble de los filamentos de actina en los podocitos.23 GEN CD2AP EL SÍNDROME NEFRÓTICO CORTICORRESISTENTE Se encontró que la alteración del CD2-AP aumenta el riesgo de glomeruloesclerosis focal y segmentaria. La GNFS es descripta como una patología con fenotipo heterogéneo . Kim et col. encontraron que en ratones con insuficiencia del gen de CD2AP exhiben un fenotipo similar a al FSGS humana. Los mismo autores en una población africana con GNFS idiopática y en 15 africanos americanos con GNFS asociada a HIV detectaron 6 distintas variantes DNA. La variante de un nucleótido, pudo ser detectada en 2 pacientes con FSGS idiopática, donde se vio la alteración de la expresión del CD2AP. Los ratones knock out CD2AP desarrollan proteinuria, inmunodeficiencia T y su muerte se produce antes de los 2 meses de vida. La proteína CD2AP en el dominio del diafragma de filtración actúa como molécula de fijación de la nefrina al citoesqueleto de actina31 LA PODOCALIXINA 7q32-q3335 Esta sialo-proteína cubre enteramente los podocitos En el modelo experimental con déficit del gen de la podocalixina los animales presentan anuria y defecto en el desarrollo y diferenciación de los podocitos. GEN TW1 11p13 EL SÍNDROME DE DENYS-DRASH: Se caracteriza por un síndrome nefrótico congénito o de comienzo temprano asociado pseudohermafroditismo XY y tumor de Wilms, sin embargo, también se ha descripto en pacientes XX.23 La lesión glomerular característica, la cual es la esclerosis mesangial difusa . La nefropatía se diagnostica generalmente al nacimiento o dentro del primer mes de vida. Se produce por mutaciones en el gen del Tumor de Wilms. Este gen está localizado en el cromosoma 11p 13, y codifica a un factor de transcripción que juega un rol crítico en el desarrollo del rińón y de las gónadas.23 Esta proteína se expresa mucho durante el desarrollo embrionario, pero en el rińón maduro su expresión persiste solo en los podocitos y en las células epiteliales de la cápsula de Bowman.23 En animales transgénicos Guo y col. demostraron que la reducción de la expresión del WT1 resultaba en glomerulonefritis con crescientes o esclerosis mesangial. Los genes de la nefrina y la podocalixina estuvieron regulados en menos en ratones con alteraciones del gen WT1 .Los autores hipotetizan que algunos pacientes con SNCR podrían tener alteraciones del WT1 y sin presentar anormalidades gonadales. Conclusiones En cuanto a la función de barrera de filtración, el diafragma de hendidura es la estructura más importante de la pared capilar glomerular. Los mecanismos que conducen hacia las anormalidades estructurales de la barrera de filtración y que llevan al síndrome nefrótico todavía están siendo dilucidados. Aparentemente son necesarios diferentes componentes estructurales para mantener la integridad de la misma. Aparte de la nefrina, recientemente se descubrieron varias proteínas intracelulares y de membrana, las cuales fueron asociadas con el desarrollo de estados proteinúricos. La podocina, una proteína de membrana específica del podocito, se encuentra mutada en varios pacientes con síndrome nefrótico esteroide resistente. Los ratones que carecen de la proteína asociada CD2 desarrollan proteinuria aproximadamente a las dos semanas de vida y mueren de fallo renal 4 a 5 semanas después. La alfa actinina cuatro es un componente del citoesqueleto de los podocitos, y es una proteína que ha sido encontrada mutada en algunos pacientes con una forma autosómica dominante de gloméruloesclerosis focal y segmentaria. La falta o la anormalidad de la función de la nefrina en el síndrome nefrótico finlandés es crucial para el desarrollo de esta enfermedad. Su ausencia lleva a la proteinuria masiva en etapas tan tempranas como la intrauterina. La angiotensina II regula la función glomerular modulando el tono arteriolar y el coeficiente de filtración Kf, con receptores presentes en el músculo liso vascular, las células mesangiales y endoteliales de los capilares. Recientemente, y como ya hemos mencionado, se descubrieron receptores para angiotensina II en el podocito. En los últimos ańos se ha acumulado información que sugiere un papel fundamental del podocito en la proteinuria y en la función del glomérulo. Además, los avances en la biología del podocito van a permitir desarrollar aproximaciones preventivas y terapéuticas destinadas a influir sobre la resistencia de esta célula a la lesión y sobre su capacidad de regeneración. La descripción de proteínas específicas de podocitos y la comprobación de que defectos de estas proteínas causan síndrome nefrótico abren la puerta para explorar el posible papel de polimorfismos de estas proteínas en la predisposición al desarrollo o a la progresión de nefropatías proteinúricas. Por otra parte, desde 1997 es posible estudiar podocitos diferenciados en cultivo, lo que permitirá abordar el efecto directo sobre esta célula de factores patogénicos de la lesión glomerular y de tratamientos existentes o potenciales. Otro campo virgen es el de la regeneración del podocito. Los datos recientes que indican la posibilidad de regeneración del miocardio lesionado a partir de células pluripotenciales derivadas de la médula ósea y la formación de nuevas neuronas en el adulto, hacen pensar que la pérdida de podocitos puede ser reversible si llegamos a comprender los mecanismos moleculares que regulan la diferenciación, desdiferenciación, proliferación y supervivencia del podocito. AGRADECIMIENTO A LA DRA ROXANA CHIRICO BIBLIOGRAFÍA
|
 3,
3, 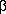 1 y la megalina.3, 11
1 y la megalina.3, 11 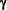 -catenina). Estas cateninas interactúan con la cadherina intracitoplasmática que las une a la actina del citoesqueleto, y traducen seńales intercelulares.15 A través de ellas, la nefrina, regularía el tamańo del poro y la permeoselectividad del diafragma.
-catenina). Estas cateninas interactúan con la cadherina intracitoplasmática que las une a la actina del citoesqueleto, y traducen seńales intercelulares.15 A través de ellas, la nefrina, regularía el tamańo del poro y la permeoselectividad del diafragma.