
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
"Inflammation and Oxidant Stress in End-Stage Renal Disease Patients"Mary Lou Wratten and Mauro AttiBellco SpA, Scientific Affairs Department, Mirandola (MO) Italymarylou.wratten@bellcospa.itAbstract End-stage renal disease patients have increased inflammation, oxidant stress and cardiovascular disease. Although there are many factors that contribute to these conditions, there also appear to be several key factors that are pivotal in these amplified processes. This presentation will discuss the role of oxidative stress, endothelial dysfunction and key signalling pathway that may be important in the progression of uremic morbidities associated with inflammation. Cardiovascular disease contributes to approximately 45% of the overall mortality of ESRD patients. While many patients have traditional risk factors such as hypertension, high frequency of diabetes, hypertriglyceridemia, low physical activity, there is also a high mortality in younger ESRD patients and patients without traditional risk factors. One of the challenges in examining this problem is that there are components related to metabolic and nutritional dysregulation, immune dysfunction and accumulation of uremic toxins. These are coupled with extracorporeal and pharmacologic treatments that, on one hand, are beneficial in attempting to replace renal function, but on the other hand, may be inadequate or even contribute to inflammation and oxidative stress. Discovering the mechanisms and interaction of the various pathways may be beneficial in developing effective prevention and treatment strategies. Introduction Uremic patients often have altered immune and inflammatory states. What isn’t always clear is if these states are due to uremia per se and underlying co-morbidities or whether inflammation or immune function can be either positively or negatively influenced by extracorporeal replacement therapies. Oxidant stress and inflammation are processes that are intricately related. Do reactive oxygen species play a role in the development of cardiovascular disease and uremic co-morbidities? ESRD patients often have increased markers of oxidative damage, decreased levels or activities of several antioxidants and several co-morbidities that are associated with free-radical mediated damage. Inflammation is a complex process that is both an epi-phenomenon and an integral process in the evolution of ESRD pathologies. Multiple oxidative processes occur as both physiologic mediators of normal cell processes and as pathological mediators if the reactive oxygen species are produced at inappropriate concentrations, at inappropriate locations or react with the wrong molecules. The localized aspect and activation of specific cellular pathways often makes evaluation of free radical-mediated damage difficult to assess. As an example, in atherosclerosis, there are often obscure microenvironments where notable biochemical, cellular and physiologic changes occur, but that cannot be seen at a systemic level. Extracorporeal treatments Biocompatibility and the treatment (therapeutic) conditions are two parameters that appear to influence oxidative stress and inflammation. In the early years of hemodialysis, treatments were performed without a lot of regard to the cellular or host response to the treatment. Treatments were performed with relatively "bioincompatible materials" and with much less emphasis on treatment optimization. Today we have an enormous range of tools (ranging from biocompatible materials, ultrapure dialysate, enhanced convective methods of toxin removal), but nevertheless co-morbidities and cardiovascular mortality are much higher than in the general population. Modern dialytic techniques are increasingly oriented towards "global biocompatibility". This includes the use of non-activating dialytic membranes, ultrapure water to avoid blood contamination by endotoxin or cytokine inducing substances, products that are free of releasable contaminants and the use of state-of-the-art machines that aid in correct dialytic delivery. New therapies are also being proposed that allow individualized biofeedback to maintain appropriate physiologic balance of parameters such as sodium, other electrolytes, acid-base and towards increasing cardiovascular stability. There are numerous conflicting studies in the literature as to the role of the "best" biocompatible membrane, treatment time, treatment dose, which is the best toxin (or group of toxins to remove). Despite all of the advances in dialytic therapies, uremic patients still have significant levels of inflammation. The question arises as to whether renal replacement therapies can positively or negatively influence inflammation and oxidative stress. Notable progress has been made in our understanding of common pathways that influence inflammation - particularly with respect to cell signaling and activation of transcription factor pathways (figure 1 and 2). A problem still exists however that the localized aspect and activation of specific cellular pathways often makes evaluation of free radical mediated damage difficult to assess. This is particularly true in complex disease processes, such as atherosclerosis, where many of the cellular and biochemical changes occur in obscure microenvironments. 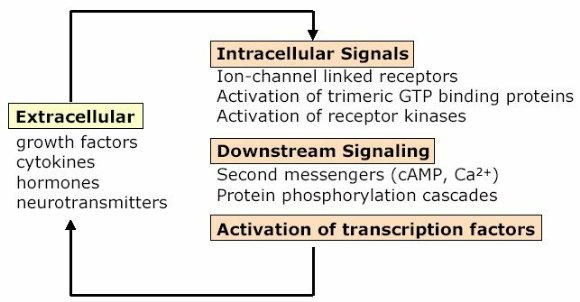 Figure 1 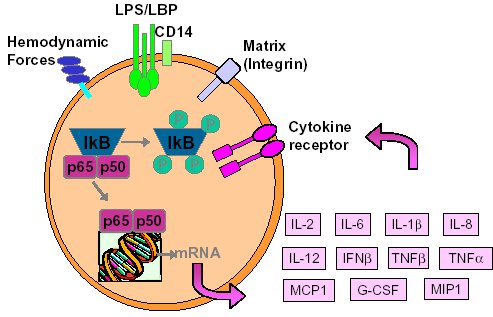 Figure 2 Intrinsic factors related to uremia can be considered as components that both aggravate and amplify the inflammatory response, as well as compensatory factors that are produced to minimize inflammation. Many hemodialysis patients have high levels of inflammatory mediators or cytokines, but the biological activity of these mediators is low due to the simultaneous presence of inhibitors. The treatment of uremia, by renal replacement therapies and pharmacologic interventions, can both add to the complications of chronic inflammation as well as play a role in reducing factors that lead to chronic inflammation. Thus, some therapies may be able to effectively reduce some of the aberrant intrinsic factors related to uremia, while others may contribute to their formation. Dialysate The quality of the water and dialysate appear to be very important in biocompatible dialysis. Dialysate patient blood is in contact with over 18,500 liters of water and is increased even further in hemodiafiltration patients. While the standards and purity of water have improved dramatically over the last 20 years, there are still many reports of water exceeding 2000cfu/ml of water borne bacteria! Even if the dialysate tests negative for endotoxin by LAL reactive substances, small fragments of gram postive or gram negative bacterial debris can pass through the dialyzer and provoke cytokine production. These cytokine inducing substances often react at toll like or other receptors and cause production of cytokines (figure 3). 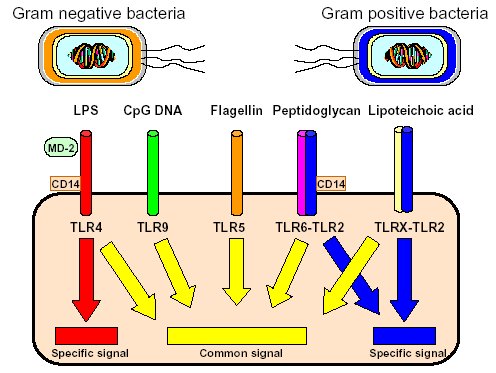 Figure 3 Dialyzers and renal replacement therapies Hemodialysis patients today have a wide choice of materials and treatment modalities. Most of the synthetic membranes are considered relatively biocompatibile with little cell activation, complement generation or thrombogenicity. A recent trend has been towards higher permeable dialyzers, treatments incorporating increased convection and the use of adsorbents. The rationale behind these treatments has been to better remove "larger uremic toxins" such as AGE-modified proteins, Patient factors: dyslipidemia ESRD patients show a secondary form of complex dyslipidemia consisting of both quantitative and qualitative abnormalities in serum lipoproteins. These are thought to result from alterations in lipoprotein metabolism and composition. The prominent features of uremic dyslipidemia are an increase in serum triglyceride levels due to elevated very low density lipoprotein (VLDL)-remnants and intermediate-density lipoprotein (IDL)) and low high-density lipoprotein (HDL) cholesterol. Low-density lipoprotein (LDL) cholesterol is often normal, but the aberrant deposition of cholesterol may originate from the atherogenic small and dense LDL subclass (sdLDL). Release of lipoprotein lipase from extrahepatic vascular surfaces during heparin administration may also be an important factor in the change in triglyceride metabolism in uremic patients. Repeated administration of heparin can lead to a lipoprotein lipase depletion - leading to decreased VLDL metabolism. Administration of heparin to hemodialysis patients may also modify many of the properties of LDL. Aggregation and fusion of LDL are thought to be important in the early events of atherogenesis. Recent evidence suggests that the glycosaminoglycan (GAG) chains of extracellular proteoglycans bind to both LDL and phospholipase A2. Both heparin treated and heparin bound lipolyzed LDL increase the tendency for the LDL particles to fuse. Oxidation of LDL could be one of the key features in the development of atherosclerosis and several authors have reported increased levels of oxidized LDL in uremic patients (figure 3). These modifications occur at the level of the apo-B100, surface phospholipids, or lipids within the core region. Oxidized low density lipoprotein (ox-LDL) has a plethora of components that are not present in native LDL and their presence and quantity depends on the nature, type, and extent of oxidation. Once modified, ox-LDL can impose an array of changes in cytokine and growth factor expression and/or release by a wide variety of cell types, and increase endothelial cell activation/dysfunction, affecting the production of important mediators such as nitric oxide. Although it is relatively easy to modify LDL in vitro, there is still a great debate over which modifications actually occur in vivo and where these modifications take place. LDL are the main carriers of cholesterol and make up a heterogeneous class of particles that can vary in size, composition and structure. They have a single copy of apo-B100. The surface monolayer of non-modified LDL is made up of approx. 700 phospholipid molecules while the core contains about 1600 cholesterol ester and 170 triglyceride molecules. In addition, LDL particles contain about 600 molecules of unesterified cholesterol that is located mostly on the particle surface. In addition to approx. 6 molecules of The subset of small dense LDL, also known as LDLIII, represents a potential risk factor for cardiovascular disease, possibly because this subpopulation is particularly susceptible to oxidation. This subclass accumulates preferentially in hypertriglyceridemic diabetic patients with nephropathy or on hemodialysis. Patient factors: Endothelial dysfunction Endothelial function is profoundly altered in uremic patients. The endothelium normally helps prevent atherosclerosis in medium to large arteries by inhibiting platelet activation and maintaining a nonproliferative and biochemically quiescent intima. It is a dynamic organ that can respond to a wide array of agonists and environmental challenges that range from specific cytokines to physical forces such as shear stress. A crucial role of the healthy endothelium is to produce a finely tuned balance of appropriate anti-clotting, anti-adhesive and vasodilatory mediators. Some of these changes in uremic patients are invariably due to responses to modified or oxidized lipoproteins, but there are many other important factors. These include shear stress, cell production of inflammatory mediators and cytokines, biochemical and enzymatic changes that promote inflammation and proliferation, as well as inhibitors and compounds that interfere with nitric oxide. Some of the effects on endothelial cell proliferation and activation appear to be moderated by oxidative pathways. For instance, oxidized LDL can stimulate NADPH oxidase and induces proliferation. There is also a strong correlation between NADPH activity, atherosclerotic risk factors and endothelial dysfunction. Both reduced endothelial vasorelaxation and increased NADPH oxidase activity are associated with increased clinical risk factors for atherosclerosis. Identification of various NADPH oxidase homologues (referred to as nox) that are cell specific and generate superoxide and H2O2 at different rates and at specific sites suggests that subtle regulation of oxidative pathways may regulate cell growth and specific cell functions. Other circulating inhibitors of endothelial function, and in particular, inhibitors of nitric oxide synthase, have also been implicated in the pathogenesis of vascular disease in chronic renal failure. Although hemodialysis can reduce the concentration of circulating inhibitors and increase flow mediated dilation, concentrations of various NOS inhibitors are often increased in uremic patients. Asymmetric dimethylarginine (ADMA) is one such inhibitor that is increased, but other inhibitors have also been proposed to be important in EDRF inhibition. Ox-LDL induces a concentration-dependent inhibition of EDRF activity that directly impairs authentic NO-induced stimulation of cGMP accumulation in detector cells. This appears to be related to the lipid component as extracted lipids from ox-LDL block NO-stimulated cGMP accumulation to about the same extent as intact ox-LDL, while the protein component of ox-LDL does not inhibit the cGMP response. Oxidized LDL may also play a role in binding to the LOX receptor with subsequent production of superoxide, which in turn inactivates nitric oxide. This may operate as part of a modulating system for induction of NO synthesis by reactive oxygen species under conditions where a large flux of superoxide is induced (as in the case of NADPH oxidase activation), eNOS may become uncoupled and thereby inactivated in terms of NO production, generating superoxide as an alternate product. Production of H2O2 by eNOS can be further aggravated in hemodialysis patients who often have low intracellular L-arginine concentrations or defects in transport and can predispose NOS to generate superoxide by uncoupled O2 reduction at its catalytic heme site. Moreover, superoxide anion (O2.-) can interact directly with NO in diffusion controlled rates, producing peroxynitrite (ONOO-). In this way, increased rates of O2.- production may divert NO from its physiological action toward pathological effects mediated by ONOO- or by the formation of other NO derived protein adducts (figure 4). 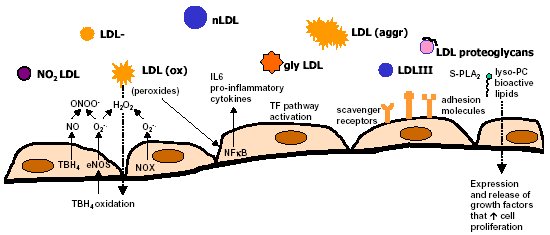 Figure 4 Conclusion Although many of the new treatments and techniques appear promising, real reduction of cardiovascular morbidity and mortality is still a distant goal. Much more basic science is needed to better define mechanisms involved in the pathogenesis of the precocious development of cardiovascular disease in renal failure patients. 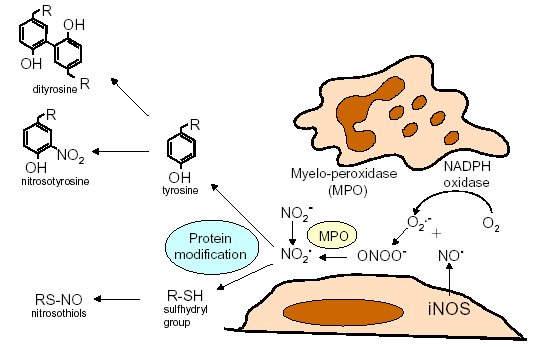 Figure 5 Selected References 1. Amore, A., and Coppo, R. Immunological basis of inflammation in dialysis. Nephrol Dial Transplant 17 Suppl 8:16-24; 2002. 2. Ando, M., Lundkvist, I., Bergstrom, J., and Lindholm, B. Enhanced scavenger receptor expression in monocyte-macrophages in dialysis patients. Kidney Int 49:773-80.; 1996. 3. Arici, M., and Walls, J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: is C-reactive protein the missing link? Kidney Int 59:407-14.; 2001. 4. Arvanitidou, M., Spaia, S., Katsinas, C., Pangidis, P., Constantinidis, T., Katsouyannopoulos, V., and Vayonas, G. Microbiological quality of water and dialysate in all haemodialysis centres of Greece. Nephrol Dial Transplant 13:949-54.; 1998. 5. Arvanitidou, M., Spaia, S., Velegraki, A., Pazarloglou, M., Kanetidis, D., Pangidis, P., Askepidis, N., Katsinas, C., Vayonas, G., and Katsouyannopoulos, V. High level of recovery of fungi from water and dialysate in haemodialysis units. J Hosp Infect 45:225-30.; 2000. 6. Baigent, C. Premature cardiovascular disease in chronic renal failure. Lancet 356:147-152; 2000. 7. Baigent, C., Burbury, K., and Wheeler, D. Premature cardiovascular disease in chronic renal failure. Lancet 356:147-52.; 2000. 8. Blackwell, T. S., and Christman, J. W. The role of nuclear factor-kappa B in cytokine gene regulation. Am J Respir Cell Mol Biol 17:3-9.; 1997. 9. Bolton, C. H., Downs, L. G., Victory, J. G., Dwight, J. F., Tomson, C. R., Mackness, M. I., and Pinkney, J. H. Endothelial dysfunction in chronic renal failure: roles of lipoprotein oxidation and pro-inflammatory cytokines. Nephrol Dial Transplant 16:1189-97.; 2001. 10. Borawski, J., Naumnik, B., Pawlak, K., and Mysliwiec, M. Endothelial dysfunction marker von Willebrand factor antigen in haemodialysis patients: associations with pre-dialysis blood pressure and the acute phase response. Nephrol Dial Transplant 16:1442-7.; 2001. 11. Cappelli, G., Perrone, S., and Ciuffreda, A. Water quality for on-line haemodiafiltration. Nephrol Dial Transplant 13:12-6.; 1998. 12. Chisolm, G., Hazen, S. L., Fox, P., and Cathcart, M. Oxidation of lipoproteins by monocytes-macrophages. J Biol Chem 274:25959-25962; 1999. 13. Chisolm, G. M., 3rd, and Chai, Y. Regulation of cell growth by oxidized LDL. Free Radic Biol Med 28:1697-707.; 2000. 14. Cominacini, L., Pasini, A. F., Garbin, U., Davoli, A., Tosetti, M. L., Campagnola, M., Rigoni, A., Pastorino, A. M., Lo Cascio, V., and Sawamura, T. Oxidized low density lipoprotein (ox-LDL) binding to ox-LDL receptor-1 in endothelial cells induces the activation of NF-kappaB through an increased production of intracellular reactive oxygen species. J Biol Chem 275:12633-8.; 2000. 15. Cominacini, L., Rigoni, A., Fratta-Pasini, A., Garbin, U., Davoli, A., Campagnola, M., Pastorino, A., Lo Cascio, V., and Sawamura, T. The binding of oxidized LDL low density lipoprotein (ox-LDL) to ox LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem 276:13750-13755; 2001. 16. Deighan, C. J., Caslake, M. J., McConnell, M., Boulton-Jones, J. M., and Packard, C. J. Atherogenic lipoprotein phenotype in end-stage renal failure: origin and extent of small dense low-density lipoprotein formation. Am J Kidney Dis 35:852-62.; 2000. 17. Hakala, J. K., Oorni, K., Ala-Korpela, M., and Kovanen, P. T. Lipolytic modification of LDL by phospholipase A2 induces particle aggregation in the absence and fusion in the presence of heparin. Arterioscler Thromb Vasc Biol 19:1276-83.; 1999. 18. Han, K., Chang, M., Boullier, A., Green, S., Li, A., Glass, C., and Quehenberger, O. Oxidized LDL reduces monocyte CCR2 expression through pathways inovlving peroxisome proliferator-activated receptor g. J Clin Invest 106:793-802; 2000. 19. Handelman, G. J., Walter, M. F., Adhikarla, R., Gross, J., Dallal, G. E., Levin, N. W., and Blumberg, J. B. Elevated plasma F2-isoprostanes in patients on long-term hemodialysis. Kidney Int 59:1960-6.; 2001. 20. Heermeier, K., Leicht, W., Palmetshofer, A., Ullrich, M., Wanner, C., and Galle, J. Oxidized LDL suppresses NF-kappaB and overcomes protection from apoptosis in activated endothelial cells. J Am Soc Nephrol 12:456-463; 2001. 21. Himmelfarb, J., and McMonagle, E. Albumin is the major plasma protein target of oxidant stress in uremia. Kidney Int 60:358-363; 2001. 22. Hurt-Camejo, E., Camejo, G., and Sartipy, P. Phospholipase A2 and small, dense low-density lipoprotein. Curr Opin Lipidol 11:465-71.; 2000. 23. Irish, A. Cardiovascular disease, fibrinogen and the acute phase response: associations with lipids and blood pressure in patients with chronic renal disease. Atherosclerosis 137:133-9.; 1998. 24. Koniger, M., Quaschning, T., Wanner, C., Schollmeyer, P., and Kramer-Guth, A. Abnormalities in lipoprotein metabolism in hemodialysis patients. Kidney Int Suppl 71:S248-50.; 1999. 25. Kovanen, P. T., and Pentikainen, M. O. Secretory group II phospholipase A(2) : a newly recognized acute-phase reactant with a role in atherogenesis. Circ Res 86:610-2.; 2000. 26. Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest 108:793-797; 2001. 27. Locatelli, F., Bommer, J., London, G., Martin-Malo, A., Wanner, C., Yaqoob, M., and Zoccali, C. Cardiovascular disease deterinants in chronic renal failure: Clinical approach and treatment. Nephrol Dial Transplant 16:459-468; 2001. 28. Lonnemann, G. Chronic inflammation in hemodialysis: the role of contaminated dialysate. Blood Purif 18:214-23.; 2000. 29. Lonnemann, G. Should ultrapure dialysate be mandatory? Nephrol Dial Transplant 15 (suppl 1):55-59; 2000. 30. Mimic-Oka, J., Simic, T., Djukanovic, L., Reljic, Z., and Davicevic, Z. Alteration in plasma antioxidant capacity in various degrees of chronic renal failure. Clin Nephrol 51:233-41.; 1999. 31. Morris, S. T., McMurray, J. J., Spiers, A., and Jardine, A. G. Impaired endothelial function in isolated human uremic resistance arteries. Kidney Int 60:1077-82.; 2001. 32. Murase, T., Kume, N., Korenaga, R., Ando, J., Sawamura, T., Masaki, T., and Kita, T. Fluid shear stress transcriptionally induces lectin-like oxidized LDL receptor-1 in vascular endothelial cells. Circ Res 83:328-33.; 1998. 33. Nguyen-Khoa, T., Massy, Z. A., De Bandt, J. P., Kebede, M., Salama, L., Lambrey, G., Witko-Sarsat, V., Drueke, T. B., Lacour, B., and Thevenin, M. Oxidative stress and haemodialysis: role of inflammation and duration of dialysis treatment. Nephrol Dial Transplant 16:335-40.; 2001. 34. Parthasarathy, S., Santanam, N., Ramachandran, S., and Meilhac, O. Oxidants and antioxidants in atherogenesis. An appraisal. J Lipid Res 40:2143-57.; 1999. 35. Penn, M. S., Patel, C. V., Cui, M. Z., DiCorleto, P. E., and Chisolm, G. M. LDL increases inactive tissue factor on vascular smooth muscle cell surfaces: hydrogen peroxide activates latent cell surface tissue factor. Circulation 99:1753-9.; 1999. 36. Petit, L., Lesnik, P., Dachet, C., Moreau, M., and Chapman, M. J. Tissue factor pathway inhibitor is expressed by human monocyte-derived macrophages : relationship to tissue factor induction by cholesterol and oxidized LDL. Arterioscler Thromb Vasc Biol 19:309-15.; 1999. 37. Prichard, S. Dyslipidemia as a risk factor for cardiac disease in dialysis patients. Seminars in Dialysis 12:87-90; 1999. 38. Quaschning, T., Krane, V., Metzger, T., and Wanner, C. Abnormalities in uremic lipoprotein metabolism and its impact on cardiovascular disease. Am J Kidney Dis 38:S14-9.; 2001. 39. Rajman, I., Harper, L., McPake, D., Kendall, M., and Wheeler, D. Low-density lipoportein subfraction profiles in chronic renal failure. Nephrol Dial Transplant 13:2281-2287; 1998. 40. Rigatto, C., and Kingal, P. Oxidative stress in uremia: Impact on cardiac disease in dialysis patients. Seminars in Dialysis 12:91-96; 1999. 41. Thannickal, F., and Fanburg, B. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279:L1005-L1028; 2000. 42. Uittenbogaard, A., Shaul, P. W., Yuhanna, I. S., Blair, A., and Smart, E. J. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem 275:11278-83.; 2000. 43. Witko-Sarsat, V., Friedlander, M., Nguyen Khoa, T., Capeillere-Blandin, C., Nguyen, A. T., Canteloup, S., Dayer, J. M., Jungers, P., Drueke, T., and Descamps-Latscha, B. Advanced oxidation protein products as novel mediators of inflammation and monocyte activation in chronic renal failure. J Immunol 161:2524-32.; 1998. 44. Wratten, M. L., Galaris, D., Tetta, C., and Sevanian, A. Evolution of oxidative stress and inflammation during hemodialysis and their contribution to cardiovascular disease. Antioxid Redox Signal 4:935-44.; 2002. 45. Yorioka, N., Taniguchi, Y., Yamashita, K., Ueda, C., Nakamura, C., Harada, S., and Yamakido, M. Tissue factor and tissue factor pathway inhibitor in hemodialysis patients. Int J Artif Organs 21:699-701.; 1998. 46. Zimmermann, J., Herrlinger, S., Pruy, A., Metzger, T., and Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int 55:648-658; 1999. |
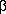 2-microglobulin, oxidized proteins, factor D and other toxins that are often not adequately removed by conventional hemodialysis. Although there is some concern that "beneficial molecules" may be lost during high efficiency treatments, other factors need to also be taken into consideration. Albumin is a molecule that is of particular concern since many hemodialysis patients have low serum albumin levels and hypoalbuminemia is associated with increased morbidity and mortality. Its loss is of understandable concern since 60-70% of hemodialysis patients have low serum albumin levels, however, since it is a negative acute phase protein, the increased removal of other proinflammatory toxins may actually be of greater benefit to reducing the acute phase response than the loss of a small percentage of albumin. It should also be noted that circulating albumin in hemodialysis patients may be compromised by the loss of its antioxidant properties (due to oxidation of its sulfhydryl group) or by saturation with uremic toxins. In this case, small losses of albumin may actually turn out to be beneficial in stimulating new protein synthesis.
2-microglobulin, oxidized proteins, factor D and other toxins that are often not adequately removed by conventional hemodialysis. Although there is some concern that "beneficial molecules" may be lost during high efficiency treatments, other factors need to also be taken into consideration. Albumin is a molecule that is of particular concern since many hemodialysis patients have low serum albumin levels and hypoalbuminemia is associated with increased morbidity and mortality. Its loss is of understandable concern since 60-70% of hemodialysis patients have low serum albumin levels, however, since it is a negative acute phase protein, the increased removal of other proinflammatory toxins may actually be of greater benefit to reducing the acute phase response than the loss of a small percentage of albumin. It should also be noted that circulating albumin in hemodialysis patients may be compromised by the loss of its antioxidant properties (due to oxidation of its sulfhydryl group) or by saturation with uremic toxins. In this case, small losses of albumin may actually turn out to be beneficial in stimulating new protein synthesis. 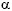 -tocopherol (vitamin E), LDL also contains
-tocopherol (vitamin E), LDL also contains 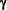 -tocopherol, carotenoids, oxycarotnenoids and ubiquinone.
-tocopherol, carotenoids, oxycarotnenoids and ubiquinone.