
|
Paneles de Discussión
Paneais de Discussio Comunicaciones libres
Comunicaçoes livres |
NOVEL CARDIOVASCULAR
RISK FACTORS IN END STAGE RENAL DISEASE
Carmine Zoccali
 In the last three decades intensive investigations have lead to a paradigm shift in the interpretation of atherosclerosis, from a purely metabolic process (i.e. mainly driven by hypercholesterolemia) to a disease where inflammation is the dominant alteration. This paradigm shift owes very much to the late Robert Ross who lucidly formulated the response to injury theory of atherosclerosis.  The response to injury theory holds that offending factors (which I will discuss later on) damage the endothelium and disrupt its physiologic properties. The adhesiveness of the endothelium to leukocytes and platelets is increased and the endothelium synthesises vasoactive molecules, cytokines, and growth factors. Macrophages, that is inflammatory cells, migrate in the arterial wall where they take up cholesterol and lipids and become foam cells. The lipid core is thus generated. A fibrous cap covers the lipid core and the plaque alters the hemodynamic properties of vessel. Plaques may ulcer thus triggering thrombosis. Thrombi detach from the ulcerated plaque and eventually occlude arterial vessels at distant districts, the heart and the brain, thus causing the most feared consequences of atherosclerosis.  In patients with ESRD systemic evidence of a micro-inflammatory process is associated with atherosclerosis because the number of atherosclerotic plaques in the carotid artery is strongly associated are evident in patients with ESRD because there is direct relationship between atherosclerotic plaques and CRP. More importantly in these patients serum CRP represents a strong predictor of adverse cardiovascular events. In a German cohort of dialysis patients the CV death rate was about 5 times higher in patients in the 4th CRP quartile than in those in the first CRP quartile and these results were fully confirmed also in an American study by Kaysen group. 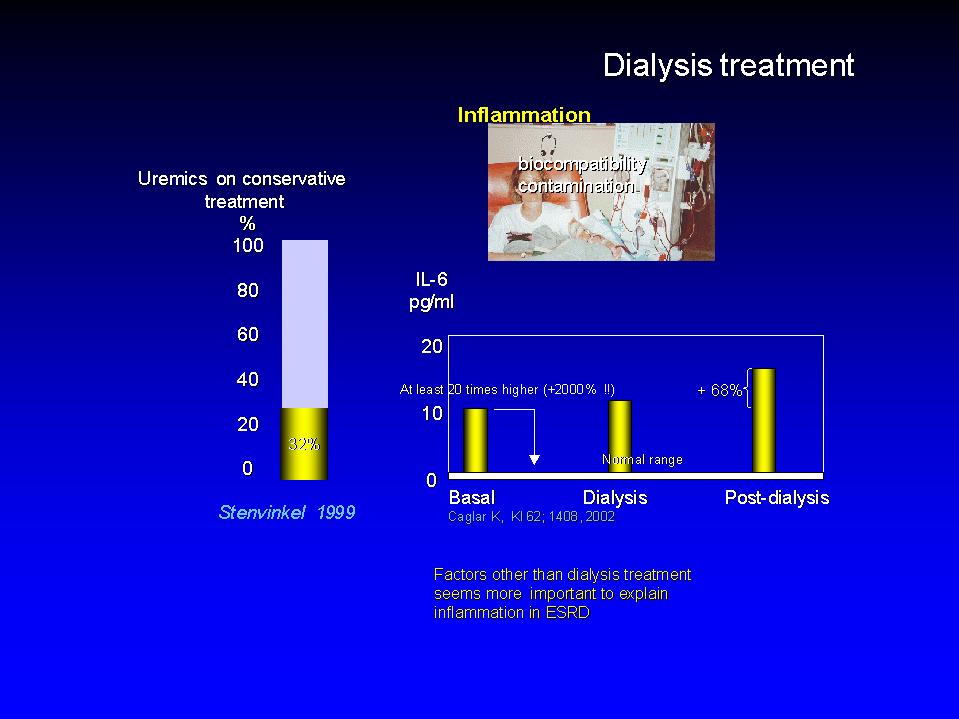 Dialysis treatment, i.e. biocompatibility and dialysate contamination have long been suspected as culprits of inflammation in ESRD. It has been shown that IL-6, an inflammation marker increases by the 68% two hours after dialysis clearly showing that dialysis per se is an inflammatory stimulus. However, IL-6 in baseline conditions was 20 times higher, 2000% higher than in healthy individuals indicating that the increase induced by dialysis is just a tiny fraction of the problem. Furthermore evidence of inflammation as measured by serum CRP is evident in about 1/3 of patients before dialysis. Overall it is reasonable to say that factors other than dialysis treatment are implicated in inflammation in ESRD.  In this presentation I will discuss how traditional and non traditional risk factors impinge upon inflammation and cardiovascular risk. In other words I will try to assemble an inflammation driven perspective of cardiovascular risk in ESRD.  Skipping Cholesterol I will touch upon three traditional risk factor, hypertension, smoking and diabetes. I will then move to factors peculiar to ESRD, i.e. hyperphosphatemia and hyperparathyroidism and dialysis treatment and finally to so called emerging risk factors, hyperhomocysteinemia, infections and endogenous factors that disturb endothelial function like ADMA.  Hypertension is a complex phenotype because causative factors implicated in hypertension in ESRD, renin angiotensin and sympathetic systems over-activity and chronic volume overload have other pressure independent effects on the CV system, pro-inflammatory and growth promoting effects.  Angiotensin II is a recognised pro-inflammatory substance and growth promoter. Double transgenic rats overexpressing the angiotensinogen and renin gene display marked LVH and the texture of the myocardium in these rats shows an important infiltration of macrophages, that is inflammatory changes. A similar process may be at work in ESRD disease because it has been shown that LV mass in these patients is directly related to PRA. The higher PRA, the higher LV mass.  The sympathetic system is a major regulator of cardiovascular function but has also many other important functions. It interferes with immune mechanism and inflammation and NE is a growth promoter of myocardial cells. Norepinephrine and sympathetic fibres are well represented in linphnodes and the sympathetic system is recognised as a modulator of the systemic response to inflammation.  On the other hand norepinephrine is a recognised growth promoter for myocardial cells and in experimental animals it induces LVH by mechanisms independent of BP. In this regard it is interesting noting that in patients with ESRD circulating NE is directly related to the muscular component of the LV, the mean ventricular wall, as well as with the prevalence of concentric hypertrophy. More importantly raised NE is predictor of cardiovascular death. Of note both these effects on the heart and on survival are independent of arterial pressure.  Chronic volume overload besides being an hypertension trigger also induces the expression of pro-inflammatory molecules in the heart. Indeed in an experimental model of volume overload Monocyte Chemoattractant Protein is overexpressed in cardiomyocites and there is evidence that in other experiments that also TNF and IL6 are over-expressed in myocardial cells when end diastolic pressure is high. It is interesting that the overexpression of MCP occurs not only in animals with decompensated heart failure but also in those that have compensated forms of the disease. Fibrinogen is an acute phase reactant of particular interest in relation to volume overload indeed it is not only responsive to inflammatory stimuli like IL-6 but also to volume stimuli.  Indeed in HD patients without systemic evidence of inflammation that also display raised plasma volume the synthesis rate and the plasma concentration of Fibrinogen is increased. In support of a volume expansion as a potential trigger of fibrinogen synthesis Kaysen has recently shown that the plasma concentration of this protein is directly related to albumin synthesis. Because the plasma concentration of Fibrinogen reflects two powerful CV-event triggers it is expected to be strongly related to CV events  In keeping with this hypothesis Fibrinogen is indeed a strong predictor of incident cardiovascular events in patients with ESRD. As you see event free survival was substantially reduced in patients with high Fibrinogen (those in the third tertile) than in those with relatively lower values and in an adjusted analysis a 200 mg/dl increase in plasma fibrinogen was associated with a 50% increase in cardiovascular risk.  Cigarette smoke is a deadly gas mixture. Smoking is a strong oxidant stimulus and has an inflammatory effect. Indeed macrophages of asymptomatic smokers produce a greater amount of inflammatory cytokines than those of healthy age and sex matched controls. The proportion of smokers is fairly variable among countries and France and Italy have been less successful than America for controlling this problem. In France and Italy the proportion of smokers in dialysis patients is almost identical to that of the general population and Jungers in a study in patients with renal failure living in the Ile de France province found that the risk of cardiovascular complications is much higher in smokers than in smokers.  Diabetics with ESRD are at extreme risk of CV events and here I will only allude to this condition. The very high risk of diabetes is very often associated with other risk factors, hypercholesterolemia and hypertension. Furthermore diabetics on dialysis have a long history of insulin resistance and the exposure to AGEs in these patients is much higher than that of other HD patients. It is well recognised that AGEs constitute an inflammatory stimulus and insulin resistance has been linked with raised CRP in the general population.  Let's now move to factors peculiar to ESRD. High Ca x P and vascular calcifications and dialysis treatment.  Vascular calcifications in the coronary, in heart valves and in peripheral arteries are pervasive in ESRD. Undoubtedly uncontrolled hyperphosphatemia favours vascular calcifications because the risk of all cause mortality is 21% higher and that of incident CV events 45% higher in patients with serum P >6.5 mg/dl. However hyperphosphatemia and hypercalcemia is just one face of the coin.  Osteoclast like cells are demonstrable in heart valves and in arterial vessels. Their ability to affect phosphate metabolism is documented by their alkaline phosphatase activity. It is interesting to note that this activity is much enhanced when these cells are co-cultured with monocyte macrophages thus providing a link between inflammation and vascular calcification. On the other hand in heart valves and arterial vessels as well there is a true process of ossification. When we have true lamellar bone, inhibitors of calcification are important, particularly so in patients with ESRD. In this regard Fetuin is a most interesting calcification inhibitor and inverse acute phase reactant. When Fetuin is low inflammation and calcification should be more likely. In agreement with this hypothesis Ketteler has recently shown that CV events free survival is shorter in patients with low Fetuin than in those with relatively higher values.  Let's now move to emerging risk factors. Homocysteine, infection and endogenous inhibitors of NO synthase  Homocysteine. Evidence has been recently provided that Homocysteine constitutes an inflammatory stimulus because in rats treated with methionine, an aminoacid that raises homocysteine , here to levels 4 times higher than normal, there is a marked in increase in the expression of MCP, ICAM and VCAM in the arterial walls of these rats that also show macrophage infiltration particularly in the luminal side.  Infection has long been suspected by microbiologists as a cardiovascular risk factor. The most implicated agents are a virus, Cytomegalovirus, and two bacteria Helycobacter and Chlamydia Pneumoniae. To be implicated as a causative agent a bacterium or a virus should pass the test of stringent criteria established almost a century ago by test Virchow. The agent should be isolated in affected individuals, an animal model should be developed, the appearance of the agent should precede the appearance of the diseases and the eradiction of the agent should be associated with the cure of the disease. Over 1000 paper investigating the relationship between infectious agents and cardiovascular risk have been published but until now there is little conclusive evidence. No agent has passed in full the test of Virchow criteria.  In patients with ESRD infections of the vascular access are an established source of occult inflammatory processes. Indeed in a survey in 115 patients in dialysis centres in the Houston area, all 20 patient presenting with fever of unknown origin, infection -purulent infection- in the graft was the cause of occult fever. Furthermore access graft infection was also demonstrated in 68% of 21 asymptomatic patients patients with occluded grafts.  Raised CRP has also been associated with periodontal infections. In fact the antibody titer IgG to Porphiromonas gengivalis and bacteriodes fortuitus is higher in patients with CRP > 10 mg/L than in those with lower values. However, the implication of these findings in relationship to CV risk remain at best elusive.  Chlamydia Pneumoniae is undoubdtedly the most implicated agent in human atherosclerosis. Here we have also an animal model. The model used by Saikku was the New Zealand rabbit. This rabbit does not develop atherosclerosis at his normal diet. Saikku submitted 9 rabbits to two nasal inoculations three months apart. The results were positive because 6 out nine rats developed obvious atherosclerotic lesions, the plaques here are directly visible.  Even more important Chlamydia has been repeatedly demonstrated in atherosclerotic lesions. In fact the probability of finding Chlamydia in atherosclerotic plaques in 18 times higher than in apparently normal arterial tissue.  As to dialysis patients, in 1998 we reported that in men on chronic dialysis IgG anti-Chlamydia titre was an independent correlate, though a slight one (p=0.03), of the number of atherosclerotic plaques. This finding was replicated in uraemic patients maintained on conservative treatment by Stenvinkel who showed that IgA anti-Chlamydia antibodies are independently related to intima media cross sectional area. But these associations are weak evidence, we ought to have more solid proof of an involvement of Chlamydia in the high mortality in dialysis patients.  We examined the problem in a cohort study involving 278 patients tested for Chlamydia at baseline, the follow up is 4 years and we have 58 CV deaths, a strong end-point. In this data base, taking as a reference group patients negative for Chlamydia, the unadjusted risk for cardiovascular death rose in parallel with Chlamydia titre. The higher the titre the higher the risk and the Hazard ratio was of 3.24 in the top titre. Again a positive association but we ought to be careful because confounding may be considerable.  Indeed the anti-Chlamydia antibody titre was related to other risk factors: directly to age, sex, smoking and diabetes and inversely to serum cholesterol suggesting that malnutrition may be a factor predisposing to Chlamydia infection.  When we adjusted the analysis for these confounders, age, sex, smoking, diabetes, cholesterol the relationship became much weaker and of slight significance. It seems unlikely that Chlamydia is a major cardiovascular risk factor in dialysis patients.  To briefly summarise. We have seen that several factors both traditional and non traditional factor may damage the arterial system in ESRD and we have also seen that most of these factor operate by enhancing cytokine production and by inducing endothelial dysfunction. But vasculotoxicity may be only a part of the problem. There may well be a defect in vasculoprotection, in other words the problem may be rooted inside the cardiovascular system.  Under normal conditions NO is continuously generated in th endothelium because the enzyme NO synthase transforms L Arginine into NO and citrulline. NO in the endothelium has a protective role for the cardiovascular system because NO inhibits vascular muscle cells proliferation, platelets aggregability and the adhesion of monocytes to the endothelium, all process that trigger atherosclerosis.  There are endogenous inhibitors of this enzyme and ADMA, asymmetric dymethylarginine is the most important. ADMA can be eliminated by the kidney but there is a very important alternative metabolic route, that is cellular metabolism by Diethyl-Diamino-Hydrolase an enzyme present within the endothelial cells which is very sensitive to oxidative stress.  In our study population plasma ADMA concentration was 3 times higher than in healthy control subjects. The median value was about 1 in healthy subjects and about 3 in dialysis patients , a 2 micromoles difference. We will comment soon on the implication of this difference in terms of CV risk. Since ADMA accumulation generates a pro-atherogenic situation we examined the relationship between ADMA and incident mortality and CV events in a cohort study in 225 HD patients.  First of all some crude analyses. Here patients are separated in two groups: survivors and non-survivors. ADMA was 3 micromoles in survivors and 2 in non-survivors, similarly it was much higher in patients who had CV events than in those who had no such events. However crude analyses may be misleading because do not take into account the influence of other risk factors. For this reason we did a proper survival analysis by the Cox's regression model.  To do survival analysis we stratified patients into 3 groups: the first groups including those with ADMA less than the 50th percentile (the median), the second including those with levels between the 50th and the 75th percentile and the third including those with values above the 75th percentile. Survival decreased dramatically from the first to the third group so that the risk of death was 2.6 times higher in those in the first group than in those in the first. Please note that the HR was fully adjusted for all significant risk factors in this population. By the same token the HR for incident CV events was 2.2 times higher in patients in the third ADMA group than in those in the first group.  Because the relationship between and ADMA and survival appeared to be linear, there is another way to extrapolate the implication of survival analysis. As you remember, there is a 2 micromole/L increase in plasma ADMA in HD patients in comparison to healthy subjects. This 2 micromoles increase is associated with a 52% higher risk of death and with a 34% higher risk of CV events.  Thus ADMA appears to be a risk factor peculiar to end stage renal disease of top importance and should be without hesitation added to the list. However we ought to keep in mind that a risk factor can be considered as a causal one only and only if it passes the test of intervention studies. We believe that the data we gathered represent a strong argument for not deferring such studies any more. ADMA is a modifiable risk factors and the hypothesis that it is a causal risk factor may undergo formal experimental testing in appropriate studies. |