|
Paneles de Discussión
Paneais Conferencias
Palestras
|
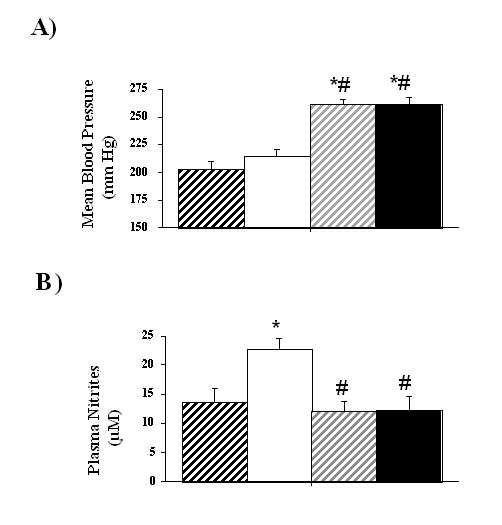
Figure 1. Changes in blood pressure (A) and plasma nitrite levels (B) at the end of the study. UNX-SHRs were treated during six months with: D-Arg (1 mg kg-1 min-1; strong hatch columns), L-Arg (1 mg kg-1 min-1; open columns) alone or in the presence of L-NAME (1 mg kg-1 min-1 the first month and 0.5 mg kg-1 min-1 the next five months; respectively light hatch and close columns). Each symbol represents the mean + SEM of at least 12 animals. *P<0.05 vs. D-Arg, # P<0.05 vs. L-Arg. Renal parameters were summarized in Table II. The creatinine clearance, urinary flow, proteinuria and sodium excretion were similar in all the treated UNX-SHRs.
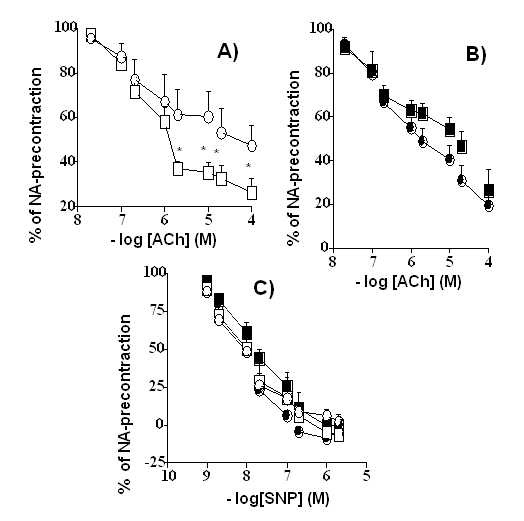
Acetylcholine-and nitroprusside-induced vasorelaxations Addition of NA (10-6 M) to rat thoracic aortic rings induced in different treated UNX-SHRs a contraction of: D-Arg = 2826 + 160 mg (n =18), L-Arg = 2396 + 210 mg (n =15), L-NAME+D-Arg = 2337+149 mg (n =18) and L-NAME+L-Arg = 2435+112 mg (n =18). Data on vascular relaxations were summarized in Figure 2 and and Table III. The response was significantly higher (p<0.01) in the D-Arg group in comparison with others. In NA (10-6M)-precontracted rings, the sensitivity and Emax in concentration-responses curves to ACh were significantly reduced in vessels of D-Arg treated-animals vs. other treated-groups. However, in NA (10-6 M)-precontracted rings, although there were differences in the sensitivity between groups in concentration-responses curves to SNP (a nitric oxide-donor) the Emax effects were similar.
Figure 2. Concentration-response curves for acetylcholine (Ach; A and B) and sodium nitroprusside (SNP; C) in throracic aorta rings of UNX-SHRs. Results are expressed as percentage dilatation after noradrenaline (10-6 M)-induced precontraction. Rats were treated during six months with: D-Arg (1 mg kg-1 min-1), L-Arg (1 mg kg-1 min-1) alone or in the presence of L-NAME (1 mg kg-1 min-1 the first month and 0.5 mg kg-1min-1 the next five months). Each symbol represents the mean + SEM of at least 6 aortic rings. *P<0.05 vs. D-Arg.
Aortic structure
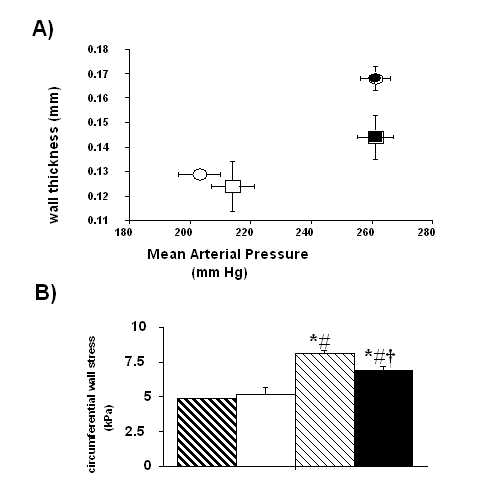
Table IV and Figure 3 show the effects of different treatments in thoracic aortic structure. The mean internal diameter was similar in all treated UNX-SHRs. However, the wall thickness, media-to-lumen ratio and media-cross-sectional area where significantly higher in the L-NAME+D-Arg group in comparison with others. It is interesting to observe some differences between L-NAME+L-Arg treated-animals vs. other groups when MAP related parameters, such as wall thickness/MAP and circumferential wall stress, were analyzed.
Figure 3. Aortic thickness/mean arterial pressure ratio (A) and circumferential wall stress (B) of UNX-SHRs. Rats were treated during six months with D-Arg (1 mg kg-1 min-1; A: open circles and B: strong hatch columns); L-Arg (1 mg kg-1 min-1; A: open squarts and B: open columns); L-NAME+D-Arg (1 mg kg-1 min-1 the first month and 0.5 mg kg-1 min-1 the next five months plus D-Arg; A: close circles and B: light hatch columns); and L-NAME+L-ARG (1 mg kg-1 min-1 during the first month and 0.5 mg kg-1 min-1 during the next five months plus L-Arg; A: close squarts and B: close columns). Each symbol represents the mean + SEM of at least four data. *P<0.05 vs. D-Arg, #P<0.05 vs. L-Arg and † P<0.05 vs. L-NAME+D-Arg. DISCUSSION In view of the kidney and blood pressure are closely related [38] and a renal defect may represent the response of the kidney to the hypertensive progress [39]; we have investigated in this study the long-term role of NO on aortic vessels in a mixed experimental process: a nephron reduction that accelerate renal lesions [40] and SHR, an animal model of essential hypertension [8-9]. According plasma nitrite levels measured at the end of the study, a chronic administration of low doses of L-NAME, were able to blunt NO-production induced by 1 mg kg-1 day-1 of L-Arg. However, L-NAME aggravates blood pressure in this model after 6 months of administration, with or without L-Arg, and indicates that NO is not the main regulator of blood pressure, after the renal resetting that occurs upon UNX in genetic hypertension. NO synthesis blockade also induced hypertrophy of the aortic wall of UNX-SHRs, but this structural change is more related on blood pressure rise than NO production. This finding is probably one of the most important ones in this work. The other interesting finding of this study is that those UNX-SHRs treated with L-NAME+L-Arg have low survival, perhaps adaptation to maintain high blood pressure makes necessary to preserve the abnormal vascular structure. Endothelial dysfunction is a typical characteristic of hypertension. It is the result of an imbalance in the normal equilibrium between endothelial contracting and relaxing factors [2, 41-43]. Both pressure-dependent and -independent factors have been implicated in its appearance [2]. Particularly in SHRs, NO production upon stimulation is normal or even elevated, but it is partially neutralized by an abnormally excessive production of oxygen reactive species [44-45]. Here, we show that neither long-term NO synthesis inhibition nor supplementation with L-Arg induces significant changes in the intrinsic relaxation capacity of the aorta (whether endothelium-dependent or -independent). This means that, in our model, the behavior of the aortic wall as regards its capacity of relaxation is not determined by the blood pressure levels and its consequences, but rather by genetic or pre-established determinants. All groups have a similar aortic relaxation profile in spite of being subject to starkly different pressure levels. When we tested vascular reactivity, we found some differences between groups on NA-induced contraction. If vascular tone and sympathetic response could be dependent on NO release [46], our data on NA-precontracted vessels are clear the consequence of these effects. In the aftermath of the present results on vasorelaxations, we can conclude that this status is not altered by the presence or absence of NO in the aortic wall. The resultant endothelial imbalance seems not to be dependent on further pressure increase or on NO status, since all groups showed a similar response to ACh, except D-Arg. The modification of other endothelium-derived factors, as a consequence of an adaptation pressure load, with a reciprocal regulation of their production with NO would be implicated on this response. In fact, two important local endothelial vasoconstrictors such as arginine II and endothelin-1 would be regulated according NO synthesis [27, 42, 47]. In additional, it is demonstrated that a vasoconstrictor factor derived from cyclooxygenase pathway is up regulated in the vascular wall of UNX-SHRs [48]. Some of these products of arachidonic acid such as 20-hydroxyeicosatetraenoic acid (20-HETE), epoxyeicosatrienoic acids (EETs) or prostaglandin H2 and tromboxane A2 may be also regulated by NO production [49-52]. It is also interesting to observe that the endothelial hyperpolarizing relaxing factor, actually considered as a derivate of EETs, is dependent on cytochrome P450 [53]. If we block NO production with L-NAME that is also able to inhibit EETs formation, because its blocks the reduction of ferric cytochrome C by ferrous ion [54], perhaps we are increasing blood pressure by two different sites. When morphometric results were analyzed in UNX-SHRs, it is interesting to observe on aortic wall a different degree of MAP with the same internal diameter, as previous reported [55-57], which could be related again to an endothelial dysfunction. Nervertheless, we can not discard a possible role of NO in vascular cell growth [58-59], according data of the L-NAME+D-ARG group on wall thickness and media cross-sectional area; we demonstrate some differences in arterial walls of large arteries as an adaptative process leading to regulate blood flow [35], when data on aortic structural changes were corrected with the MAP. Rat aortic structure has been studied extensively in hypertension in SHRs [60, for review see reference 61] and NO deficiency using high doses of L-NAME [62], but not one has linked essential hypertension with renal mass reduction, chronic NO inhibition and structural changes in large arteries for a long time. Although, our structural changes in aortas of UNX-SHRs were not so large, because doses of different NO drugs were very low; its evidences that NO could modify the adaptative process to hypertension of large arteries by means of a low dose of L-ARG; in fact, L-NAME+L-ARG treated animals had a significant reduction in circumferential wall stress and wall thickness/MAP in comparison with L-NAME + D-ARG group and this could explain the different survival of the animals. Finally, according our data there are not significant differences in renal function among groups. All groups of UNX-SHRs, regardless of the treatment they have received, reset the remaining kidney to achieve normal renal performance. However, those animals with their NO-pathway blocked by L-NAME administration, needed higher blood pressure to maintain both a normal renal function and a balance in sodium excretion, in agreement with the well-known role of NO on the relationship between blood pressure/intraglomerular pressure and natriuresis/diuresis [63-65]. It is interesting to observe that L-NAME did not inhibit L-Arg effects. Perhaps, L-Arg transport into cells might be altered or reset after UNX, as previous reported in humans with renal damage [66], and the main control of renal function would not depend on NO actions. In conclusion we could suggest in UNX-SHRs treated during 6 months with low doses of NO related drugs that: a) renal dysfunction may be an independent process from vascular response and vascular reactivity; and b) functional and structural changes in thoracic aorta seems to be linked to both pressure load, and changes in NO production.
ACKNOWLEDGMENTS The authors thank Luis Muñoz de la Pascua for expert technical assistance and his assistants: Juan Villoria Terrón and José Fernando Martín Martín.
REFERENCES
1 Cain AE, Khalil RA. Pathophysiology of essential hypertension: role of the pump, the vessel, and the kidney. Semin Nephrol 2002; 22: 3-16.
2 Spieker LE, Noll G, Ruschitzka FT, Maier W, Lüscher TF. Working under pressure: the vascular endothelium in arterial hypertension. J Hum Hypertens 2000; 14: 617-630.
3 Packer CS. Changes in arterial smooth muscle contractility, contractile proteins and arterial wall structure in spontaneous hypertension. Proc Soc Exp Biol Med 207: 148-174.
4 Marín J. Mechanisms involved in the increased vascular resistance in hypertension. J Autonom Pharmacol 1993; 13: 127-176.
5 Neutel JM. Beyond the sphyngmomanometric numbers: hypertension as a syndrome. Am J Hypertens 2001; 14(pt 2): 250S-257S.
6 Taddei S, Virdis A, Ghiadoni L, Salvetti G, Salvetti A. Endothelial dysfunction in hypertension. J Nephrol 2001; 13: 205-210.
7 Lee MRKW. Vascular changes at the prehypertensive phase in the mesenteric arteries from the spontaneously hypertensive rats. Blood Vessels 1983; 22: 105-126.
8 Okamoto K. Spontaneous hypertension in rats. Int. Rev. Exp. Pathol. 1969; 7: 227-270.
9 Trippodo NC, Frohlich ED. Similarities of genetic (spontaneous) hypertension: man and rat. Circ Res 1981; 48: 309-319.
10 Dworkin LD, Freiner HD. Glomerular injury in uninephrectomized spontaneously hypertensive rats. Aconsequence of glomerular capillary hypertension. J Clin Invest 1986; 77: 797-809.
11 Huang W, Lee SL, Sjoquist M, Effects of neurohypophyseal antagonists in postnephrectomy natriuresis in male rats. Kidney Int 1994; 45: 692-699.
12 Palmer RMJ, Ashton DS, Moncada S. Vascular endothelium cells synthetize NO from L-arginine. Nature 1988; 333: 664-666.
13 Schini-Kerth VB, Vanhoútte PM. Nitric oxide synthases in vascular cells. Exp Physiol 1995; 80: 885-905.
14 Loscalzo J. What we know and don´t know about l-arginine and NO. Circulation 2000; 101: 2126-2133.
15 Bachmann S, Mundel P. Nitric oxide in the kidney: synthesis, localization and function. Am. J. Kidney Dis 1994; 24: 112-129.
16 Reyes AA, Karl IE, Klahr S. Role of arginine in health and renal disease. Am J Physiol 1994; 267: F331-F346.
17 Furchgott RF, Zawaski JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288: 373-376.
18 Baylis C, Harton P, Engels K. Endothelial derived relaxing factor (EDRF) controls renal hemodynamics in the normal rat kidney. J Am Soc Nephrol 1990; 1: 875-881.
19 Majid DS, Navar LG. Nitric oxide in the control of renal hemodynamics and excretory function. Am. J. Hypertens. 2001; 14(6 Pt 2): 74S-82S.
20 Cooke JP., Dzau VJ. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med 1997; 48: 489-509.
21 Kone BC, Baylis C. Biosynthesis and homeostatic roles of nitric oxide in the normal kidney. Am J. Physiol. 1997; 272: F561-F578.
22 Winquist RJ, Bunting PB, Baskin EP, Wallance AA. Decreased endothelium-dependent relaxation in New Zealand genetic hypertensive rats. J Hypertens 1984; 2: 541-545.
23 Pallone TL, Mattson DL. Role of nitric oxide in regulation of renal medulla in normal and hypertensive kidneys. Curr Opin Nephrol Hypertens 2002; 11: 93-98.
24 Ribeiro MO, Antunes E, deNucci G, Lovisolo SM Zatz R. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension 1992; 20: 298-303.
25 Zats R, Baylis C. Chronic nitric oxide inhibition model six years on. Hypertension 1998; 32 958-964.
26 Ono H, Ono Y, Takanohashi A, Matsuoka H, Frohlich ED. Apoptosis and glomerular injury after prolonged nitric oxide synthase inhibition in spontaneously hypertensive rats. Hypertension 2001; 38: 1300-1306.
27 Ono H, Ono Y, Frohlich ED. ACE inhibition prevents and reversed L-NAME exhacerbated nephrosclerosis in spontaneously hypertensive rats. Hypertension 1996; 27: 176-183.
28 Reverte M, Flores O, Gallego B, Lestón A, López-Novoa JM. Effect of chronic NG-nitro-L-arginine methyl ester (L-NAME) on blood pressure and renal function in conscious uninephrectomized spontaneously hypertensive rats. Can J Physiol Pharmacol 1998; 76: 63-67.
29 Weder AB, Schork NJ. Adaptation, allometry, and hypertension. Hypertension 1994; 24: 145-156.
30 Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996; 271: C1424-1437.
31 Calver A, Collier J, Vallance P. Dilator actions of arginine in human peripheral vasculature. Clin. Sci. 1991; 81: 695-700.
32 Kauser K, Rubanyi GM. Gender differences in endothelial dysfunction in the aorta of spontaneously hypertensive rats. Hypertension 1995; 25(part 1): 517-523.
33 Pfeffer JM, Pfeffer MA, Frohlich ED. Validity of an indirect tail-cuff method for determining systolic arterial pressure in unanesthetized normotensive and spontaneously hypertensive rats. J Lab Clin Med 1971; 78: 957-962.
34 Griess P. Bermerkukungen zu der Abhandlung der HH. Wesley and Benedikt ueber einige Azoverbindungen. Ber Dtsch Chem Ges 1879; 12: 426.
35 Mourad J-J, Girerd X, Boutouyrie P, Safar M, Laurent S. Opposite effects of remodeling and hypertrophy in arterial compliance in hypertension. Hypertension 1998; 31(part 2): 529-533.
36 Jaffé M. Über den Niederschlag, welchen Pikrinsäure in normalen Harn erzeugt und über eine neue Reaktion des Kreatinins. Hoppe-Seyler´s Z. physiol. Chem. 1886; 10: 391-400.
37 Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Annal Biochem 1976; 72: 248-254.
38 Ruilope LM, Campo C, Rodríguez-Artalejo F, Lahera V, García-Robles R, Rodicio JL. Blood pressure and renal function: therapeutic implications. J Hypertens 1996; 14: 1259-1263.
39 Folkow B. Kidneys in primary hypertension-initiators, stabilizers or/and victim aggravators. Blood Pressure 1994; 3: 212-215.
40 Rodríguez-López A, Flórez O, Arévalo M, López-Novoa M. Glomerular cell proliferation and apoptosis in the early phase of renal damage in uninephrectomized spontaneously hypertensive rats. Kidney Int 1998; 54 (suppl. 68): S36-S40.
41 Puddu P, Puddu GM, Zaca F, Muscari A. Endothelial dysfunction in hypertension. Acta Cardiol 2000; 55: 221-232.
42 Tadei S, Virdis A, Ghiadoni L, Salvetti A. Vascular effects of endothelin-1 in essential hypertension: relationship with ciclooxygenase-derived endothelium-dependent contracting factors and nitric-oxide. J cardiovasc Pharmacol 2000; 35 (4 suppl 2): S37-S40.
43 Shimokawa H. Endothelial dysfunction in hypertension. J Atheroscler Thromb 4: 118-127
44 Varizi ND, Ni Z, Oveisi F, Trnavsky-Hobbs DL. Effect of antioxidant therapy on blood pressure and NO synthase expression in Hypertensive rats. Hypertension 2000; 36: 957-964.
45 Nava E, Lüscher TF. Evidence for an increase production of nitric oxide in spontaneously hypertensive rats. Eur J Clin Invest 1994; 24(suppl.2): A264.
46 Vargas HM, Ignarro LJ, Chaudhuri G. Physiological release of nitric oxide is dependent on the level of vasculature tone. Eur. J. Pharmacol. 1990; 190: 393-397.
47 Li J-S, Deng LY, Grove K, Deschepper CF, Schiffrin EL. Comparison of effect of endothelin antagonism and angiotensin-converting enzyme inhibition on blood pressure and vascular structure in spontaneously hypertensive rats treated with N-nitro-L-arginine methyl ester: correlation with topography and vascular endothelin-1 gene expression. Hypertension 1996; 28: 188-195.
48 López-Hernández FJ, Montero MJ, Carrón R. Mesenteric cyclooxygenase products after combined antihypertensive treatment in uninephrectomized SHRs. Cardiovasc Drugs Ther 2000; 14: 41-48.
49 Wink DA, Osawa Y, Darbyshine JF, Jones CR, Eshenaus SC, Nims R.W. Inhibition of cytochrome P-450 by nitric oxide and a nitric oxide releasing agent. Arch. Biochem. Biophys 1993; 300: 115-123.
50 Katseuko OG, Gross SS, Rifkind AB, Vane JR. Nitric oxide is a mediator of the decrease in cytochrome P450-dependent metabolism caused by immunostimulants. Proc Natl Acad Sci USA 1993; 90: 11147-11151.
51 Huang A, Koller A. Both nitric oxide and prostaglandin-mediated responses are impaired in skeletal muscle arterioles of hypertensive rats. J Hypertens 1996; 14: 887-895.
52 Shiokoshi T, Ohsaki Y, Kawabe J, Fujino T, Kikuchi K. Downregulation of nitric oxide accumulation by cyclooxigenase-2 induction and thromboxane A2 production in interleukin-1beta-stimulated rat aortic smooth muscle cells. J Hypertens 2002; 20: 455-61.
53 Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 2002; 82: 131-85.
54 Peterson D., Peterson DC, Archer S, Weir EK. The non specific nitric oxyde synthase inhibitors. Biochem Biophys Res Commu. 1992; 187: 797-801.
55 Raij L. Nitric oxide in hypertension: relationship with renal injury and left ventricular hypertrophy. Hypertension 1998; 31(part2): 189-193.
56 Hughes AD, Schachter M. Hypertension and blood vessels. Br Med Bull 1994; 50: 356-370.
57 Rizzoni D, Porteri E, Castellano M, Bettoni G, Muiesan ML, Tiberio G, Giulini SM, Rossi G, Bernini G, Agabiti-Rosei E. Endothelial dysfunction in hypertension is independent from the etiology and from vascular structure. Hypertension 1998; 31(part 2): 335-341.
58 Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromocyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 1989; 83: 1774-1777.
59 Sarkar R, Gordon D, Stanley JC, Webb RC. Cell cycle effects of nitric oxide on vascular smooth muscle cells. Am J Physiol 1997; 272: H1810-H1818.
60 Laurent S, Vanhoutte P, Cavero I, Chabrier PE, Dupuis B, Elghozi JL, Hamon G, Janiak P, Juillet Y, Kher A, Koen R, Madonna O, Maffarand, JP, Pruneau D, Thuillez C. The arterial wall: a new pharmacological and therapeutic target. Fundamen Clin Pharmacol 1996; 10: 243-257.
61 Safar M, Chamiot-Clerc P, Dagher G, Renaud JF. Pulse pressure, endothelium function, and arterial stiffness in spontaneously hypertensive rats. Hypertension 2001; 38: 1416-1421.
62 Bernátova I, Pechánova O, Krislék K. Mechanism of structural remodelling of the rat aorta during long-term NG-Nitro-L-arginine methyl ester treatment. Jpn J Pharmacol 1999; 81: 99-106.
63 Granger JP, Alexander BT. Abnormal pressure-natriuresis in hypertension: role of nitric oxide. Acta Physiol Scand 2000; 168: 161-168.
64 Fortepiani LA, Rodrigo E, Ortiz MC, Cachofeiro V, Atucha NM, Ruilope LM, Lahera V, García-Estan J. Pressure natriuresis in nitric oxide-deficient hypertensive rats: effect of antihypertensive treatments. J Am Soc Nephrol 1999; 10: 21-27.
65 Guarasci GR, Kline RL. Pressure natriuresis following acute and chronic inhibition of nitric oxide synthase in rats. Am J Physiol 270: R469-R478.
66 Mendes Ribeiro AC, Brunini TM, Ellory JC, Mann GE. Abnormalities in L-arginine transport and nitric oxide biosynthesis in chronic renal and heart failure. Cardiovasc Res 2001; 49: 697-712. Correspondence to:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||