PARTES BLANDAS, neurofibromatosis, tumor maligno de nervio periférico, retroperitoneo, mediastino.
Los tumores malignos del nervio periférico (TMNP) representan un 5% de los tumores malignos de partes blandas. Un 50-60% de los TMNP aparecen en pacientes con neurofibromatosis tipo 1 (1). En la presente comunicación se describen dos casos con esta asociación.
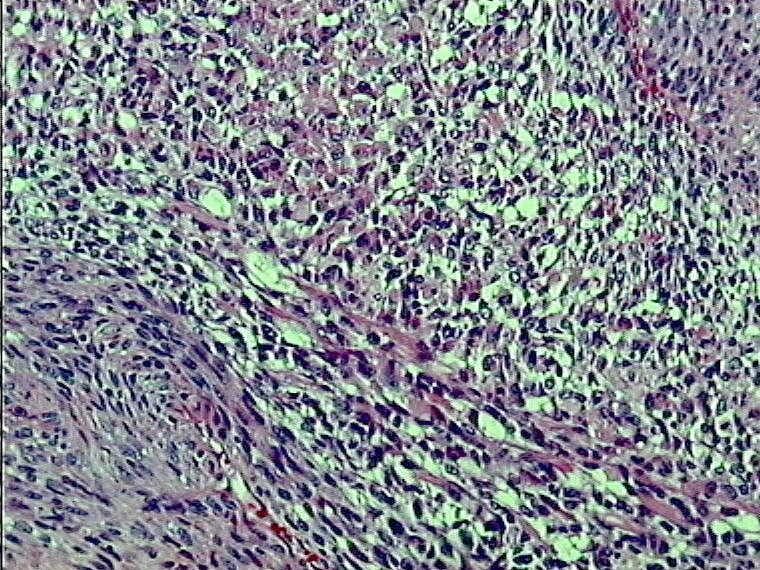
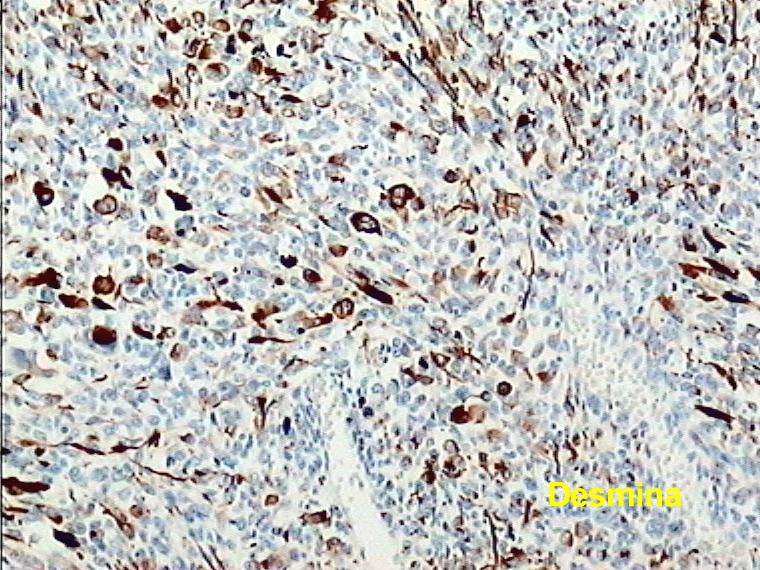
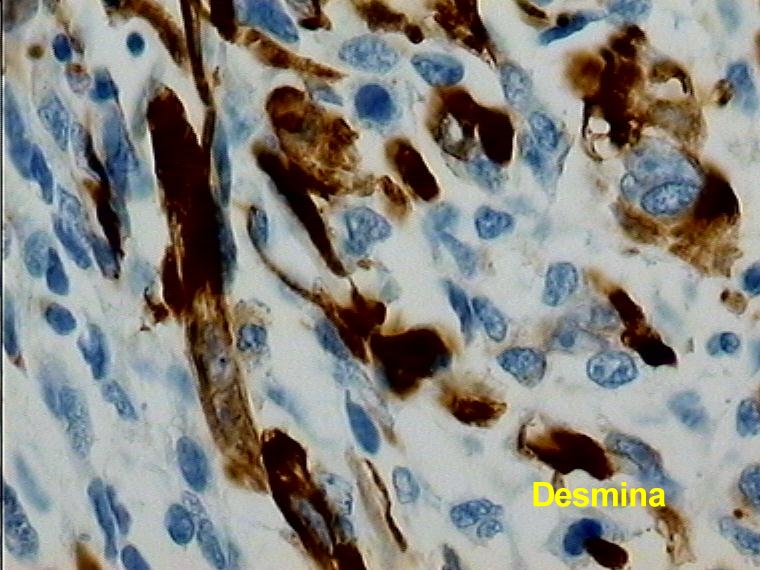
Caso 1.- Mujer de 25 años con neurofibromatosis tipo 1, con tumor de brusca aparición en mediastino superior con extensión a región cervical (figura 1). Mediante PAAF fue diagnosticado de tumor fusocelular de probable origen neurógeno. El examen macroscópico de la pieza mostró una tumoración pseudoencapsulada de 14 cm de diámetro mayor con una superficie de corte fasciculada con focos quísticos y necrohemorrágicos (figura 2). Las características microscópicas correspondían a un sarcoma fusocelular de alto grado (Figura 3). Destaba la presencia de focos con diferenciación rabdomiosarcomatosa (figura 4), con positividad intensa para desmina (figura 5 y 6) y condrosarcomatosa (figura 7).
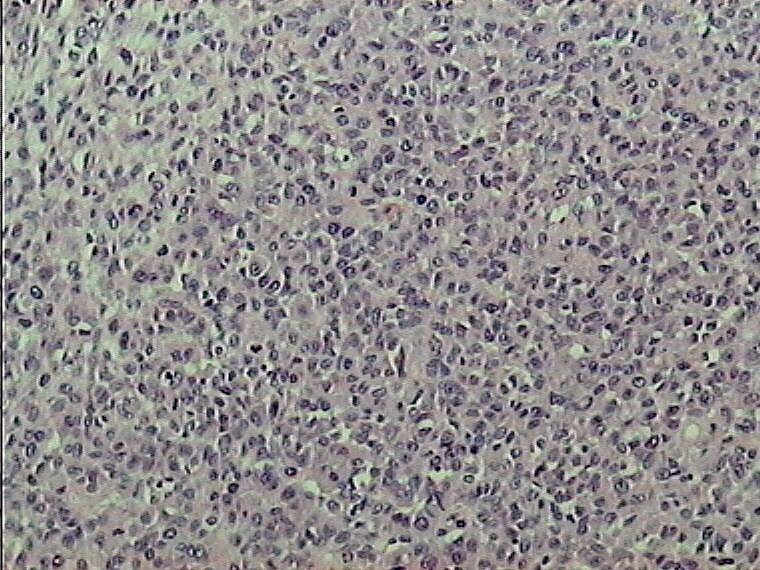
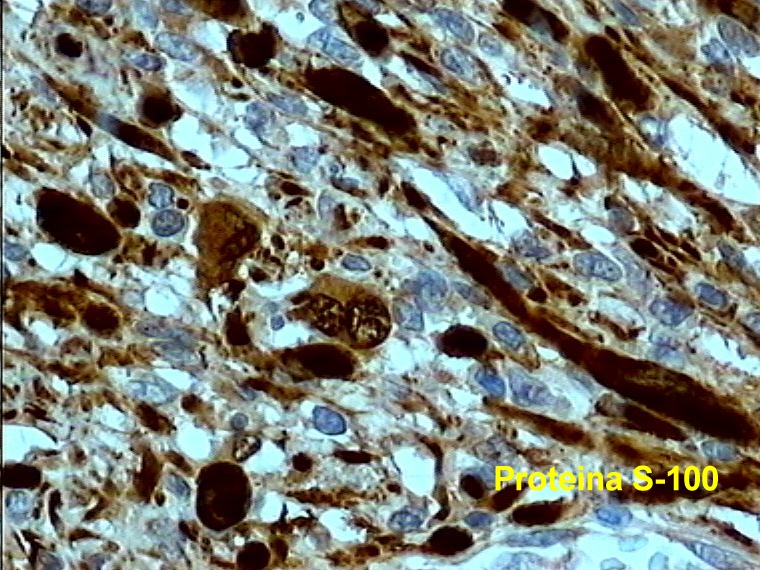
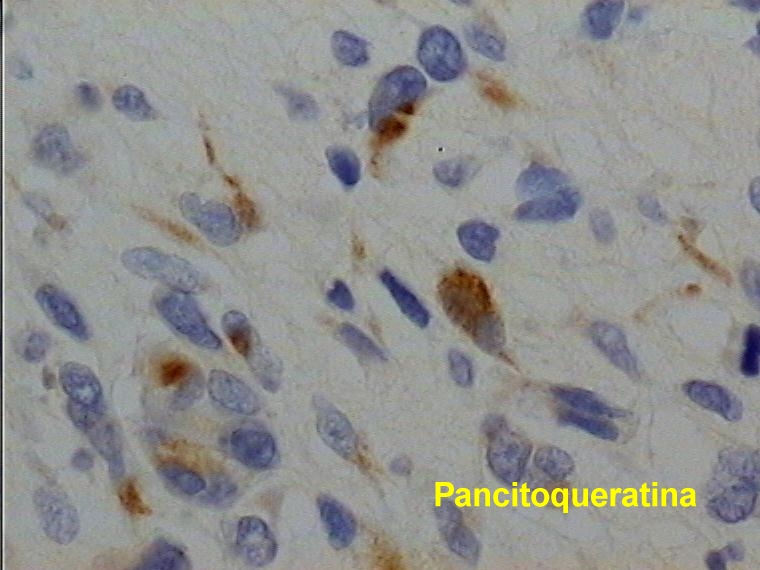
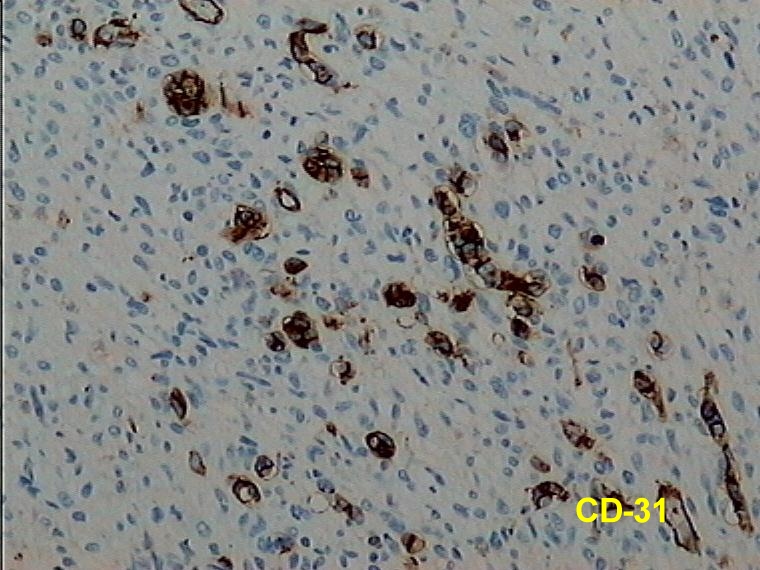
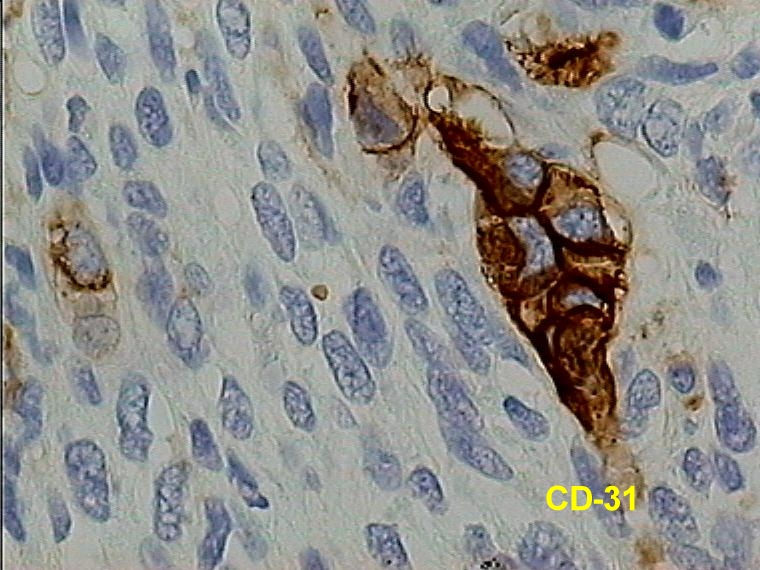
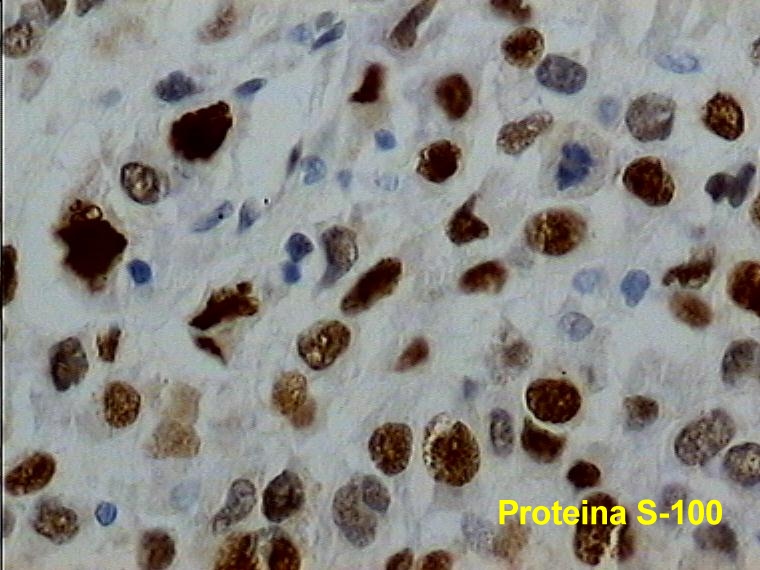
Caso 2.- Mujer de 52 años con neurofibromas múltiples y gran masa retroperitoneal. Macroscópicamente se trataba de un tumor pseudoencapsulado de 15 cm de diámetro mayor con focos de necrosis y hemorragia. El estudio microscópico mostró neurofibromas plexiformes (figura 8) con TMNP de bajo grado (figura 9) y focos de sarcoma de alto grado (figura 10). En áreas el tumor era más indiferenciado con aspecto epitelioide. El estudio inmunohistoquímico demostró positividad de las células neoplásicas para vimentina y proteína S-100 (figura 11), con positividad focal para desmina, y pancitoqueratina (figura 12) y CD31 en las áreas más epitelioides (figura 13 y 14) que sugieren diferenciación angiosarcomatosa. La proteina p53 fue positiva especialmente en las áreas mas pleomórficas, en un 80 % de las células neoplasicas (figura 15)
La mayor parte de los TMNP se originan en neurofibromas o directamente de nervios periféricos normales (3). Los criterios aceptados para el diagnóstico de TMNP son: a) Que el tumor se origine en un nervio periférico; b) Que se observe transición con un tumor de nervio periférico (neurofibroma, schwannoma, ganglioneuroma/ ganglioneuroblastoma o feocromocitoma); c) Que se desarrolle en pacientes con neurofibromatosis tipo 1, y presenten las características morfológicas habituales de los TMNP; d) Tumores desarrollados en pacientes sin neurofibromatosis tipo 1 y que presentan características morfológicas típicas de TMNP y presentan diferenciación schwanniana o perineural con métodos inmunohistoquímicos o ultraestructurales (3). La mayor parte de los TMNP se presentan en pacientes entre 20 y 50 años. Un 50-60 % de los casos se desarrollan en pacientes con neurofibromatosis tipo 1, como ocurre en los dos casos motivo de la comunicación. La presencia de elementos heterólogos ha sido descrita hasta en un 20% de los TMNP (2). La mayoría de estos elementos son histológicamente malignos e incluyen rabdomiosarcoma, condrosarcoma y osteosarcoma. El componente angiosarcomatoso en el TMNP es excepcional habiéndose descrito únicamente en 13 casos en pacientes con neurofibromatosis tipo 1 (3).