|
|
Neurofibroma con glándulas mucoproductoras. Presentación de un caso.
Autores:
Dra. Teresa Cuesta*; Dra. Bárbara Estupiñán**; Dra. Sirced Salazar***
* Servicio
de Anatomía Patológica. Hospital Clínico Quirúrgico "Hnos. Ameijeiras". País: Cuba. Email: telepatol@hha.sld.cuResumen Introducción Caso Clínico Discusión Referencias Enviar Comentarios
La mayoría de los tumores de vaina nerviosa periférica con presencia de glándulas, corresponden a tumores malignos asociados a enfermedad de von Recklinghausen. Caso clínico. Paciente masculino de 39 años de edad que consulta por tumor en el dedo de la mano derecha, no doloroso, de crecimiento lento sin antecedentes de neurofibromatosis. El estudio histológico corresponde a un tumor de vaina nerviosa periférica benigno tipo Neurofibroma, con presencia de glándulas. Las técnicas inmunohistoquímicas confirmaron la naturaleza schwannomatosa del estroma en el que se destacan elementos epiteliales glandulares bien diferenciados. El Neurofibroma con glándulas está considerado un tipo raro de diferenciación divergente resultando de gran interés para el patólogo por la necesidad de diferenciarlos de otras lesiones tumorales, siendo de gran utilidad para ello el estudio inmunohistoquímico.
La mayoría de los tumores de vaina nerviosa periférica con presencia de glándulas, corresponden a tumores malignos [ 1-8] asociados a enfermedad de von Recklinghausen [ 1,3] . Considerados un tipo raro de diferenciación divergente [ 1,4,9] resultan de interés para el patólogo por: 1) la necesidad de distinguirlos de otros tumores de tejidos blandos, como el sarcoma sinovial bifásico y el tumor de vaina nerviosa epitelioide [ 3,4,7] y 2) el reconocimiento de variantes maligna y benigna, bien establecidas, esta última aun menos frecuente [ 9-13] . Nosotros describimos aquí un tumor de vaina nerviosa periférica, con características de neurofibroma y presencia de glándulas bien desarrolladas, en focos múltiples, no asociado a neurofibromatosis.
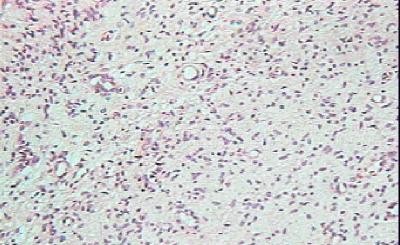
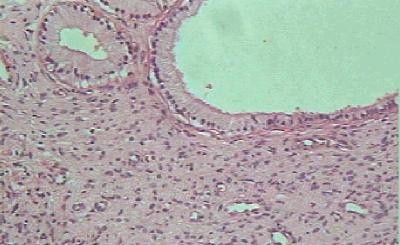
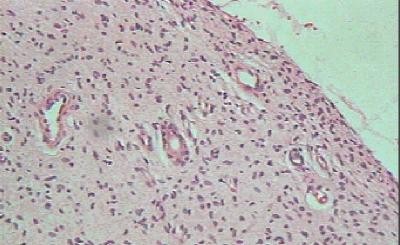
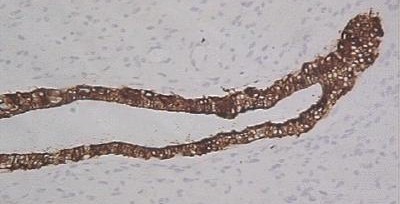
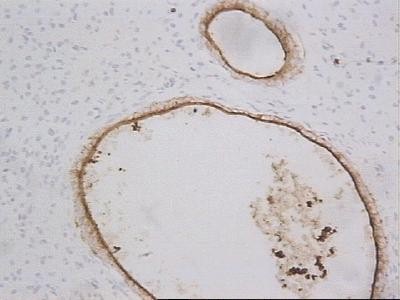
CASO CLINICO Paciente masculino, de 39 años de edad, que consulta por lesión nodular, en el borde lateral del cuarto dedo de la mano derecha, de 2.5 cm de diámetro, polilobulada, firme, movible, no adherida a piel ni planos profundos, no dolorosa con referencia de lento crecimiento hasta alcanzar su tamaño actual. No existen antecedentes familiares de neurofibromatosis. Hallazgos radiológicos: Se constata discreta radiopacidad, de contorno regular, policíclico, con integridad de estructuras óseas falángicas adyacentes. Hallazgos anatomopatológicos: Macroscópicamente, la lesión de aspecto nodular, polilobulada, contorno regular, sin cápsula definida, y consistencia firme, de 2.5 cm en su diámetro mayor, color pardo claro, ligeramente translúcido. Histológicamente, se distingue un componente fusocelular predominante (Figura 1) con orientación serpiginosa, paralela de las células que descansan en una matriz mixoide, laxa, ricamente vascularizada, con presencia de estructuras glandulares en pequeños grupos y aisladas, revestidas por epitelio simple cúbico o cilíndrico no ciliado, mucosecretor (Figura 2) que ocasionalmente se distienden adoptando configuración quística con aplanamiento del epitelio. Las células epiteliales con abundante citoplasma pálido y núcleo de disposición basal o intermedia descansan sobre membrana basal guardando orientación regular. En algunas áreas las glándulas de aspecto abortivo, se encuentran menos definidas con luces pequeñas (Figura 3). No se observaron mitosis en ninguna de las fases del tumor lo que unido a la orientación regular y detalles citomorfológicos conforman criterios de benignidad para ambos componentes.
Componente fusocelular predominante. H/E X 40
Figura 2. Glándulas revestidas de epitelio cúbico o cilíndrico no ciliado, mucosecretor. H/E X 400.
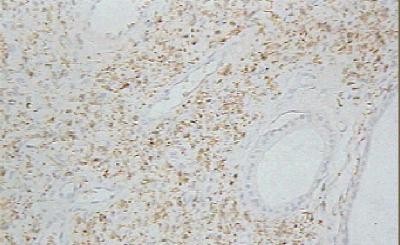
Figura 3. Glándulas abortivas con luces pequeñas. H/E X 400. La técnica de ácido periódico de Schiff (PAS) previa digestión con diastasa, destaca la secreción intraluminal y resulta positiva en parte del epitelio glandular. Desde el punto de vista inmunohistoquímico, el componente fusocelular resultó positivo para la proteína S100 (Figura 4) y la vimentina, mientras el epitelio glandular se tiñó intensamente de color pardo oscuro con la citoqueratina (AE 1/3), (Figura 5) y el antígeno de membrana epitelial (EMA). La mayoría de las células epiteliales resultaron inmunoreactivas además para el antígeno carcinoembrionario (CEA) (Figura 6).
Proteína S100 positiva en componente fusocelular. PAP X 400.
Figura 5. Citoqueratina positiva en epitelio glandular. PAP X 400
CEA positivo en epitelio glandular. PAP X 400. Los tumores de vaina nerviosa periférica glandulares han sido descritos en un rango de edades que oscila entre 8 y 89 años [ 1,2] aunque la mayoría de los casos reportados se mueven entre la cuarta y quinta décadas de la vida [ 9,10,11,14] . Woodruff y col. [ 1] , revisando once casos vistos en su laboratorio no encontraron predominio sexual, sin embargo nosotros encontramos predominio del sexo femenino en la literatura consultada [ 2,9,10,11,14] .. Los tumores de nervios periféricos con diferenciación glandular benignos son generalmente pequeños, superficiales y no suelen asociarse a neurofibromatosis [ 2,9,10] aunque Bigorgne [ 11] en 1992 reporta un caso benigno asociado a neurofibromatosis, se localizan preferentemente en el tejido celular subcutáneo de áreas diversas que incluyen tronco y extremidades igual que otros tumores de vaina nerviosa [ 12] . Pueden presentarse asociados o no a dolor [ 11,14] . Los hallazgos histológicos e inmunohistoquímicos de nuestro caso coinciden con lo descrito por otros autores [ 9,10,11,13] especialmente con el caso reportado por Oda [ 10] : la presencia de componentes bien definidos, uno fusocelular con características de neurilemoma o neurofibroma y otro epitelial con formación de estructuras glandulares, el primero positivo para la proteína S100 y la vimentina y el segundo positivo para la citoqueratina, el EMA y el CEA. Nosotros no intentamos probar diferenciación neuroendocrina en el componente epitelial, lo que ya ha sido demostrado de forma independiente por Christensen [ 15] , Karabela [ 3] y Oda [ 10] . Tras la referencia de Enzinger [ 12] del primer caso de neurofibroma benigno con presencia de glándulas mucosecretoras, Brooks [ 9] , Bigorgne [ 11] , Yoshida [ 13] y Oda [ 10] , han presentado casos semejantes, aunque sólo Enzinger [ 12] y Oda [ 10] describen neurofibromas, por lo que el caso que exponemos constituirá probablemente, el tercero reportado con estas características. La histogénesis de estos tumores aun controversial [ 4,11] ha sido atribuida a cambios metaplásicos del componente schwanniano [ 9,10] precursores de células de Schwann, células primitivas de la cresta neural [ 5] o simple atrapamiento de anejos cutáneos como afirma Woodruff [ 1] y niega Brooks [ 9] . El pronóstico de los tumores de vaina nerviosa con diferenciación glandular, depende de su naturaleza maligna o benigna, siendo los primeros los más frecuentes, cabe reconocer que sigan un curso generalmente fatal, guardando relación principalmente con el aspecto del componente fusocelular [ 4,6] más que con la presencia y características del elemento glandular, que ha reunido criterios de malignidad excepcionalmente [ 1] . Se ha recomendado en el tratamiento la extirpación quirúrgica seguida de radiación o quimioterapia para los tumores malignos [ 1] mientras los benignos curan con extirpación local total, sin requerir terapia coadyuvante, conducta seguida en este paciente, libre de enfermedad en el momento actual aunque con muy corta evolución postquirúrgica (seis meses). Sólo la extirpación incompleta explicaría recidivas en los casos benignos.
1.. Woodruff JM, Christensen WN. Glandular peripheral nerve sheath tumors. Cancer 1993; 72 (12 ): 3618-3628 . 2.. Takahara O, Nakayama Y. Malignant neurofibroma with glandular differentiation (glandular schwannoma ) . Acta Pathol Jpn 1979; 29 (4): 597-606. 3.. Karabela - Bouropoulou V, Antoniou D, Liapi - Avgeri G. Malignant peripheral sheath tumor with glandular differentiation. Report of a case with emphasis to the usefulness of inmunohistochemistry in the differential diagnosis. Arch Anat Cytol Pathol 1996; 44 ( 5-6 ): 263-268. 4.. Robbins P, Papadimitriou J. Glandular peripheral nerve sheath tumours. Pathol Res Pract 1994; 190 (4): 412-415. 5.. Wong SY, Teh M, Tan YO, Best PV. Malignant glandular triton tumor. Cancer 1991; 67( 4): 1076-1083. 6.. Woodruff JM. Peripheral nerve tumors showing glandular differentiation (glandular schwannoma). Cancer 1976; 37: 2399-2413. 7.. Sangueza OP, Requena L. Neoplasm’s with neural differentiation: a review. Part II: Malignant neoplasms. Am J Dermatopathol 1998; 20(1) : 89-102. 8.. Ferry JA, Dickersin GR. Pseudoglandular schwannoma. Am J Clin Pathol 1988; 89 (4):546-552. 9.. Brooks JJ, Draffen RM. Benign glandular schwannoma . Arch Pathol Lab Med 1992; 116 (2): 192-195. 10.. Oda Y, Hashimoto H, Tsuneyoshi M, Iwata Y. Benign glandular peripheral nerve sheath tumor. A case report. Pathol Res Pract 1994; 190 (5) 466-473. 11.. Bigorgne C, Thomine E, Hemet J, Lauret P. Benign glandular schwannoma and Recklinghausen disease. Report of a case. Ann Pathol 1992; 12 (2) : 114-120. 12.. Enzinger FM , Weiss SW. Soft Tissue Tumors. St. Louis. Toronto. London: De. Mosby, 1983. p 580-655. 13.. Yoshida SO, Toot BV. Benign glandular schwannoma. Am J Clin Pathol 1993; 100(2):167-170. 14.. Elston DM , Bergfeld WF, Biscotti CV, McMahon JT. Schwannoma with sweat duct differentiation. J Cutan Pathol 1993; 20 (3) : 254 - 258. 15.. Christensen W N, Strong E W , Bains M S , Woodruff J M . Neuroendocrine differentiation in the glandular peripheral nerve sheath tumor. Pathologic distinction from the biphasic synovial sarcoma with glands. Am J Surg Pathol 1988; 12 ( 6) : 417-426. |
Si desea contactar directamente
a los autores hágalo por email a: telepatol@hha.sld.cu |
|