|
|
Meningitis crónica basal como manifestación de sarcomatosis meníngea primaria.
Autor: Serrano Castro, Pedro J; Roig, J*; Aguilar Castillo, MŞ J**; Olivares Romero, J; Guardado Santervás, P. Sección de Neurología * Servicio de Anatomía Patológica. ** Servicio de Análisis Clínicos. Email: pserrano@meditex.es Institución: Hospital Torrecárdenas de Almería. ESPAŃA. Resumen Introducción Presentación Discusión y Conclusiones Bibliografia Enviar Comentarios RESUMEN La Sarcomatosis leptomeníngea primaria (SLP) es la neoplasia primaria intracraneal más infrecuente (0,12% del total). Se origina en las estructuras leptomeníngeas, a las que infiltra, pero sin conformar una masa tumoral identificable y su evolución suele ser fulminante. Presentamos un caso de SLP atendido en nuestro Servicio con estudio necrópsico.PRESENTACIÓN DEL CASO: Paciente de 46 ańos de edad, con AP de coronariopatía y tabaquismo que comenzó con cefalea holocraneal progresiva y ańadió un trastorno del comportamiento con agitación y agresividad, sin cortejo infeccioso. Fue atendido en salud mental. A los 2 meses de evolución el enfermo desarrolló hidrocefalia aguda por bloqueo basal que obligó a colocar una válvula de derivación ventrículo-peritoneal. El enfermo aparecía encefalopático, con tendencia al sueńo, agitación eventual y actividad alucinatoria y con severa rigidez nucal. El estudio de LCR mostró una meningitis crónica con hiperproteinorraquia severa, xantocromía y consumo de glucosa, máxima por neuroimagen en cisternas basales y con estudios etiológicos de meningitis crónicas infructuosos. El deterioro fue progresivo, desarrollando en las últimas semanas una severísima multirradiculopatía con amiotrofias y fasciculaciones. Exitus 4 meses tras el inicio del cuadro. Presentamos las imágenes histológicas macro y microscópicas del estudio necrópsico realizado a la muerte del paciente. Creemos que la SLP debe considerarse como un diagnóstico posible en casos de meningitis crónica con consumo de glucosa de mala evolución en los que se descarten otras causas más frecuentes. INTRODUCCION Actualmente se considera que los sarcomas intracraneales primarios constituyen entre el 1,9 y el 3% de todos los tumores intracraneales. Se trata de tumores compuestos por células derivadas de elementos del tejido conectivo, como fibroblastos, rabdomiocitos, lipocitos, osteoblastos o fibras musculares lisas. Probablemente esta cifra esté, por otro lado, sobrestimada, ya que se sabe que un número de otros tumores cerebrales han sido clasificados como sarcomas cerebrales en la literatura. Entre ellos cabe destacar el llamado sarcoma de células reticulares, que hoy se clasifica como un linfoma maligno primario o el hemangiopericitoma de leptomeninges, que hoy se considera una variante de meningioma angioblástico. La Sarcomatosis leptomeníngea primaria (SLP) constituye el subtipo más infrecuente dentro de los sarcomas intracraneales, representando un 10% de los mismos1. En la serie del Instituto Neurológico de Viena se encontraron 5 casos en un total de 6066 autopsias neuropatológicas2 (0,08% de las autopsias). Se trata de un tumor maligno que se origina en las células conectivas de las cubiertas meníngeas y crece en los espacios leptomeníngeos sin conformar masas circunscritas evidenciables. En la mayoría de los casos el crecimiento es focal, pero de forma excepcional puede producirse de forma difusa por las leptomeninges. Cuando esto ocurre, la expresión clínica puede ser diversa. Budka et2 al describieron 3 patrones clínicos bien diferenciados: Existen casos con sintomatología solapada entre los distintos grupos. En general la SLP es una enfermedad que aparece en 3 grupos de edad: infancia, adultos jóvenes y entre la 4Ş y 5Ş década de la vida que parecen correlacionarse con los grupos clínicos, de forma que los nińos tienden a presentar la forma pseudotumoral, los adultos jóvenes las formas mixtas y espinales progresivas y los adultos mayores la forma polirradiculoneurítica. La evolución en los casos reportados en la literatura es a la muerte en un plazo que oscila entre los 2 y los 4 meses. PRESENTACIÓN DEL CASO CLÍNICO Paciente varón de 46 ańos de edad, con antecedentes personales de vasculopatía sistémica no inflamatoria tipo Tromboangeítis obliterante tratada con cirugía de revascularización sobre arteria iliaca derecha y cardiopatía isquémica pendiente de estudio coronariográfico. Fumador activo de larga evolución. Marmolista de profesión. Durante el mes de noviembre de 1997 comenzó con una cefalea descrita como holocraneal, progresiva, acentuada durante el descanso nocturno y modificada por la movilización cefálica y las maniobras de Valsalva. No se acompańaba de nauseas o vómitos, pero sí de episodios transitorios mal descritos de diplopia eventual. Un TAC craneal practicado en dicha fecha fue informado como normal. Aproximadamente 1 mes después del inicio de la cefalea, el enfermo comenzó a presentar trastornos del comportamiento, con episodios de agresividad inmotivados. Durante este periodo huyó de su domicilio y fue encontrado a varios kilómetros sin que él pudiera dar una explicación aceptable de su comportamiento. Fue evaluado por un equipo de psiquiatría; con medicación sedante, la sintomatología pareció mejorar. Sin embargo, el día 23 de enero, durante el citado ingreso en la unidad de psiquiatría, el enfermo presentó un deterioro súbito del nivel de conciencia. Un TAC craneal practicado con carácter urgente mostró una hidrocefalia comunicante con signos radiológicos de actividad. Se decidió colocar una derivación ventrículo-peritoneal. Tras dicho procedimiento quirúrgico, el paciente pasa a planta de Neurología para investigación etiológica. En ese momento, la exploración sistémica mostraba un severo deterioro del estado general, con importante grado de desnutrición. No se visualizaban lesiones dérmicas de relevancia patológica. No existían linfadenopatías y el resto de la exploración sistémica se consideró como normal. El enfermo se encontraba consciente, aunque con severa desorientación témporo-espacial y el estado mental era fluctuante, evidenciándose en algunos momentos seguimiento ocular ficticio y autocoloquio, sugerente de actividad alucinatoria. Cuando se entablaba contacto verbal con el paciente, el lenguaje parecía correcto; en concreto, no se evidenciaban déficits disfásicos sensitivos o motores. Se apreciaba una midriasis arreactiva de ojo derecho, con pupila izquierda normal. La motilidad ocular extrínseca también era normal aunque, también de forma intermitente aparecía una divergencia ocular por exoforia de ojo izquierdo. No había asimetrías faciales ni debilidad velopalatina o lingual. El fondo de ojo mostraba papilas de bordes bien definidos sin signos retinopáticos ni estasis papilar (después de la derivación ventrículo-peritoneal). Había una discreta rigidez nucal. No se evidenciaban paresias apendiculares y los reflejos miotáticos en ese momento eran simétricos con ambos RCP flexores. Consignamos los resultados más relevantes de las pruebas complementarias practicadas:
El segundo estudio de LCR se realizó después de 10 días de tratamiento tuberculostático. En los dos casos los estudios microbiológicos (cultivos y serologías múltiples para bacterias, micobacterias y hongos) y citológicos fueron negativos.
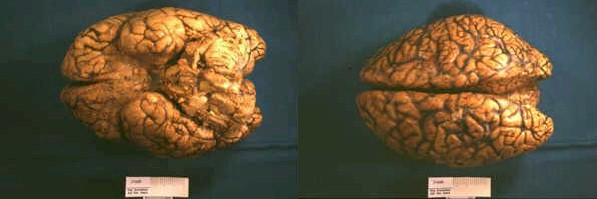
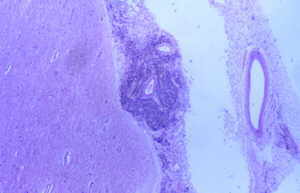
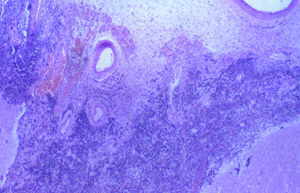
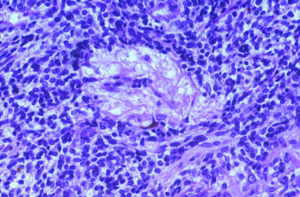
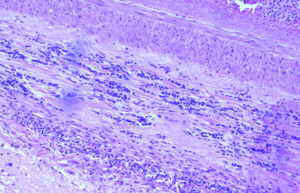
El día 30 de enero, tras el resultado del LCR se decidió iniciar tratamiento tuberculostático con 4 fármacos asociado a su tratamiento antibiótico empírico. La evolución en los días y semanas siguientes fue al empeoramiento progresivo, con deterioro del nivel de conciencia y apareciendo de forma oscilante una disconjugación ocular con abducción del ojo derecho y midriasis pupilar arreactiva ipsilateral transitoria. El enfermo, al final de la evolución clínica, presentaba una situación de estupor con ausencia de respuestas a estímulos verbales y dolorosos y era evidente una severísima polirradiculopatía que determinaba una situación de tetraparesia severa fláccida con gran componente amiotrófico y fasciculaciones espontáneas en los 4 miembros. Se repitió en varias ocasiones el estudio citológico del LCR, sin que hubiera positividad alguna. Finalmente, se produjo el exitus el día 5/3/98. La familia accedió al estudio necrópsico. En el análisis anatomopatológico del cerebro llamaba la atención: El examen macroscópico ya evidenciaba la existencia de un engrosamiento y opacificación de las leptomeninges, máximo en la porción basal de los lóbulos frontales, temporales, mesencéfalo, pedúnculos cerebrales y hemisferios cerebelosos, aunque con diseminación lateral hacia lóbulos temporales y parietales (ver Figura 1). La duramadre permanecía indemne. En el examen microscópico el dato fundamental era una proliferación neoplásica difusa por todo el lecho leptomeníngeo (Figura 2) que se infiltraba por los espacios de Virchow-Robin, a veces con infiltración focal de la sustancia gris subyacente (Figura 3).Análisis citológico de las células neoplásicas: se trataba de células de morfología fusiforme, núcleo ovoideo y picnótico con escaso citoplasma. No existía importante componente fibrótico (Figura 4). En definitiva, se trataba de células de aspecto claramente indiferenciado. Eventualmente, las células adoptaban una disposición "en fila india", simulando las típicas del meduloblastoma desmoplásico (Figura 5).
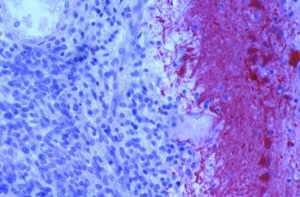
Se realizó un estudio inmunohistoquímico que incluyó el estudio de inmunorreactividad para los marcadores PS-100 y HMB-45 (marcadores para melanoma primario o metastásico), Proteína ácida glial fibrilar (GFAP, gliomatosis meníngea), NSE y sinaptofisina (meduloblastoma desmoplásico) y EMA (meningioma maligno). En la Figura 6 se puede apreciar la negatividad inmunohistoquímica para el GFAP, con intensa positividad de los astrocitos de la vecindad. Se realizó también cultivo de las estructuras meníngeas para bacterias, hongos y micobacterias, sin resultado positivo. El estudio necrópsico concluyó con la realización de un rastreo de neoplasias ocultas universal con resultado negativo. DISCUSIÓN Y CONCLUSIONES La expresión clínica de la SLP no difiere, en el análisis de los casos descritos en la literatura1-6 de la esperable en casos de infiltración neoplásica difusa de las leptomeninges, que ha sido estudiada con profundidad en infiltraciones leucémicas o carcinomatosas y que responde a la triada clásica compuesta por:
En los casos de SLP, como ya fue mencionado en la introducción se han distinguido 3 grandes categorías clínicas: polineuropática, cerebral pseudotumoral y espinal dependiendo del sitio inicial de proliferación neoplásica, pudiendo existir casos que mezclen síntomas dependientes de varias localizaciones. Nuestro caso se clasificaría como una forma inicialmente cerebral, pero con la particularidad de su tropismo por las meninges basales. En estadíos mas avanzados, la clínica mezclaba datos cerebrales con polirradiculopáticos. El dato clínico más llamativo en nuestro enfermo fue el desarrollo precoz de una hidrocefalia comunicante atribuida a un bloqueo basal de la circulación licuoral, manifestación que, a nuestro conocimiento, sólo ha sido reportada en casos de SLP en una ocasión previamente6. Esta particularidad clínica acarreó su inclusión sindrómica inicial dentro del grupo de las meningitis crónicas basales con consumo de glucosa y predeterminó el planteamiento de diagnóstico diferencial, que aparece recogido en la siguiente tabla, modificada de Ellner et al7:
Las pruebas complementarias y la misma evolución clínica en nuestro enfermo descartaron cualquiera de las causas infecciosas mencionadas, aunque en el caso de la meningitis tuberculosa, obligó a la realización de un ensayo terapéutico empírico. De las causas no infecciosas, el principal problema de diagnóstico diferencial venía dado por la posibilidad de una hemorragia subaracnoidea crónica origen indeterminado. Esta sospecha diagnóstica se veía reforzada por los antecedentes de vasculopatía conocidos en el enfermo, la aparición de una afectación completa del III par craneal izquierdo a lo largo de la evolución clínica, así como por la xantocromía evidente en las punciones lumbares repetidas. Existen casos descritos de meningitis química e hidrocefalia secundaria a bloqueo basal por sangrados subaracnoideos crónicos o repetidos. Sin embargo, la negatividad del estudio angiográfico así como de la RM espinal alejó esta posibilidad. Una dato clínico adicional que dominó en buena parte la clínica de este enfermo fue la existencia de una severa afectación encefalopática difusa desde los primeros estadíos (antes de la aparición de la hidrocefalia) así como de una clara afectación electroencefalográfica. El estudio necrópsico demostró la existencia de una afectación focal de la corteza subyacente por infiltración neoplásica (figura 3) que podría constituir el sustrato patológico de la citada encefalopatía. Descartadas las casusas de meningoencefalitis crónicas basales, con excepción de las meningitis neoplásicas, la labor diagnóstica se centró en el despistaje clínico de estos procesos. Sin embargo, los estudios citológicos fueron reiteradamente negativos y, por otro lado, la búsqueda incruenta de neoplasias ocultas resultó infructuosa. Existen 3 cuadros clínicos neoplásicos primarios del sistema nervioso central, todos ellos de presentación excepcional que pueden cursar con un perfil clínico similar al de la SLP:
En la tabla siguiente mostramos algunas características clínicas que pueden hacer sospechar uno u otro diagnóstico.
Sin embargo el diagnóstico definitivo recae en el estudio anatomopatológico. Sus principales características histológicas, según la revisión de Budka et al2 son las siguientes:
El resultado del estudio histológico de nuestro enfermo reunía los criterios esbozados por Budka et al, pudiendose clasificar dentro del subgrupo indiferenciado (Figura 4). En conclusión:
2.- Budka, H; Pilz, P and Guseo, A: "Primary leptomeningeal sarcomatosis. Clinicopathological report of six cases". J. Neurol. 1975; 211:77-93 3.- Bronfman, S; Reumont, M: "La sarcomatose méningée primitive (etude clinique et histopatologique)". J. Belge Neurol Psychiat 1947; 47:729-757. (Referenciado por Budka et al en J. Neurol, 1975;211:88). 4.- Garbes, AD: "Cytologic presentation of primary leptomeningeal sarcomatosis". Acta cytol. 1984;28:709-712. 5.- Pfluger, T; Weil, S; Weis, S et al: "MRI of primary meningeal sarcomas in two children: differential diagnostic consideration". Neuroradiology 1997; 39:225-228. 6.- Thibodeau, LL; Ariza, A and Piepmeier, JM: "Primary leptomeningeal sarcomatosis". J. Neurosurg. 1988; 68:802-805. 7.- Ellner, JJ and Bennett, JE: "Chronic meningitis". Medicine (Baltimore) 1976; 55:341. 8.- Li, CY; Witzig, TE; Phyliky, RL et al: "Diagnosis of B-cell non-Hodgkin's lymphoma of the central nervous system by inmunocytochemical analysis of cerebrospinal fluid lymphocytes". Cancer 1986; 57:737. 9.- Khang-Loon, H; Hoschner, JA and Wolfe, DE: "Primary leptomeningeal gliomatosis. Symptoms suggestive of meningitis". Arch. Neurol. 1981;38:662-666. |
Si desea contactar directamente
a los autores, hágalo por email a: pserrano@meditex.es |
|