|
| |
|
Revista
Electrónica de Medicina Intensiva
Artículo nº C35 Vol 5 nº 2, febrero 2005.
Autor: Jaime Latour Pérez
|
[ Anterior ] [ Arriba ] [ Siguiente ]
|
|
Los ensayos clínicos
aleatorizados (ECA) son la mejor herramienta con la que contamos para
evaluar la eficacia de una intervención sanitaria. El caso de la sepsis no
es una excepción; sin embargo, la historia de los ensayos ECA en
pacientes con sepsis está repleta de controversias cuando no de verdaderas
decepciones [1]. Baste recordar el caso de las megadosis de corticoides en
el shock séptico [2],
ampliamente utilizados en una época y abandonados en la actualidad, el
famoso caso de los anticuerpos anti-endotoxina
[3],
que nunca llegaron a utilizarse en la clínica, o la plétora de ensayos
fracasados con inhibidores de la cascada inflamatoria [4, 5].
Las causas de estos
fracasos se encuentran frecuentemente en la complejidad de los modelos
fisiopatológicos de la sepsis y en la dificultad de extrapolar los
resultados de la investigación animal a pacientes reales [4]. Otras veces,
sin embargo, el problema estriba en un diseño o un análisis inadecuado del
ensayo [6].
En este capítulo
pretendemos revisar los aspectos esenciales que afectan a la validez y
aplicabilidad de los resultados de los ensayos clínicos, con especial
énfasis en los ensayos realizados en pacientes con sepsis. El lector
familiarizado con el diseño de ensayos clínicos en general puede obviar la
siguiente sección y pasar directamente a la sección 3.
|
2. Estructura
básica de un ECA |
El conocimiento de la
estructura básica de un ECA ayuda a identificar los puntos vulnerables del
diseño que pueden comprometer la validez del mismo. Partiremos de la
estructura de un ECA paralelo (figura 1).
El primer paso consiste en
definir de forma precisa la pregunta de investigación. Una pregunta bien
formulada contiene al menos 3 componentes, claramente identificables: la
intervención (el tratamiento activo que se va a aplicar), la población en
la que se va a aplicar (por ejemplo pacientes con shock séptico o con
sepsis meningocócica), y el desenlace clínico con el que vamos a decidir
si la intervención realmente funciona (por ejemplo, mortalidad a los 28
días o necesidad de ventilación mecánica). Estos 3 componentes se
identifican fácilmente con el acrónimo PIO (Paciente / Intervención / Outcome
–desenlace clínico en inglés).
Figura 1
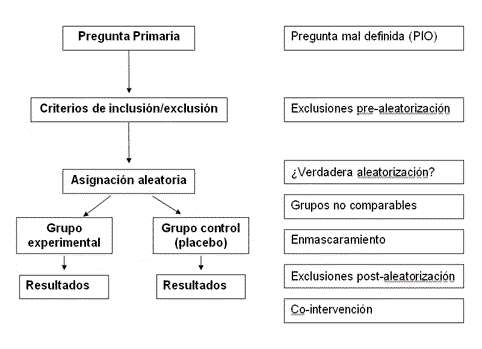
El siguiente paso consiste
en seleccionar una población de pacientes adecuada para responder a la
pregunta de investigación, mediante la definición de unos criterios
estrictos de inclusión y exclusión. Por ejemplo, si el desenlace elegido
es la mortalidad a largo plazo, no sería adecuado incluir pacientes
moribundos, que complicarían la interpretación de los resultados. De forma
similar, si el fármaco en estudio aumenta el riesgo de sufrir hemorragias,
no sería ético incluir pacientes propensos al sangrado.
La existencia de estos
criterios de inclusión y exclusión hace que la población de pacientes no
sea una muestra representativa, lo que puede limitar la
validez externa
del estudio. Por ejemplo, la exclusión de pacientes ancianos o diabéticos
puede comprometer la capacidad de generalizar los resultados a estos
grupos de pacientes. Estas exclusiones, realizadas antes de la
aleatorización, no comprometen, sin embargo la
validez interna
del estudio.
A los pacientes que cumplen
criterios de inclusión -y que aceptan participar en el estudio- se les
asigna de forma aleatoria a recibir el tratamiento activo (grupo
experimental) o a no recibirlo (grupo control). Gracias al azar, si el
número de pacientes es suficientemente amplio, se espera que los factores
pronósticos (tanto conocidos como desconocidos) se distribuyan de forma
equilibrada entre los 2 grupos, haciéndolos comparables. Sin embargo,
cuando el número de pacientes incluido en el ECA es reducido, es frecuente
que se produzcan desequilibrios entre los dos grupos. Ello obliga a
sopesar en qué medida el efecto detectado se debe al tratamiento recibido
o al diferente perfil pronóstico de los 2 grupos. A efectos de asegurar
dicha comparabilidad, y dado que un tratamiento sin actividad terapéutica
intrínseca puede tener un efecto (efecto placebo), el grupo control suele
recibir un fármaco inactivo (habitualmente añadido al tratamiento
habitual). En este caso, se dice que el ECA es controlado con placebo.
La aleatorización es el
punto clave del ECA, ya que previene el sesgo de selección, que aparece de
forma sistemática en los estudios observacionales. En éstos últimos, el
grupo de expuestos y no expuestos no son comparables en cuanto a factores
pronósticos, por lo que resulta difícil decidir -incluso con el uso de
técnicas estadísticas sofisticadas- qué parte de las diferencias
observadas se deben al tratamiento y qué parte son, sencillamente, el
resultado de las diferencias de partida entre el grupo experimental y el
grupo control.
Para que exista verdadera
aleatorización, el investigador debe ser incapaz de adivinar si el
siguiente paciente iría asignado al grupo experimental o al grupo control.
De esta forma se pretende que los criterios de inclusión-exclusión se
apliquen de la misma manera en todos los pacientes, sin favorecer
inconscientemente a ninguno de ellos. Ello se consigue mediante la
ocultación de la secuencia de aleatorización (OSA), por ejemplo mediante
el uso de sobres opacos sellados o mediante el establecimiento de una
central de aleatorización que indica al investigador (telefónicamente o
por un sistema informático) el lote que debe usar en cada paciente
individual. Diversos estudios han demostrado que los ECA que no informan
del método con el que se ha ocultado de la secuencia de aleatorización
sobreestiman el efecto en aproximadamente un 30% [7, 8].
Una vez aleatorizados, los
pacientes reciben el tratamiento activo o el control. Siempre que sea
posible, el tratamiento activo y el placebo tienen el mismo aspecto, de
forma que el grupo al que pertenece el paciente queda oculto, tanto para
el investigador como para el paciente (“doble ciego”). Con este
enmascaramiento se pretende prevenir tanto el sesgo de medición (el efecto
se mide igual en los dos grupos) como la cointervención (los dos grupos
reciben el mismo tratamiento al margen del tratamiento experimental).
Es importante no confundir
la OSA (concealled randomization)
con el enmascaramiento (blinding).
La OSA actúa previamente a la aleatorización y previene el sesgo de
selección, mientras que el enmascaramiento previene el sesgo de medición
(y la cointervención). Diversos estudios sugieren que el impacto del doble
ciego es menor que el de la OSA, de forma que, en su conjunto, los
estudios que no son “doble ciego” sobreestiman el efecto en alrededor del
15% [7, 8].
Una vez aleatorizados los
pacientes, pueden presentarse problemas que dificultan la interpretación
del estudio. Por ejemplo, puede que los pacientes del grupo experimental
no reciban realmente el tratamiento (mal cumplimiento), o que los
pacientes del grupo control reciban el tratamiento experimental
(contaminación). Otras veces se incluye (inapropiadamente) en el estudio a
pacientes que realmente no cumplían criterios de inclusión (violación del
protocolo). Asimismo es posible que los pacientes se pierdan en el
seguimiento, lo que nos impide medir de forma adecuada la frecuencia del
desenlace clínico.
A diferencia de las
exclusiones “pre-aleatorización”, las exclusiones “post-aleatorización”
pueden comprometer la validez interna del estudio. Por ejemplo, los
pacientes no cumplidores suelen presentar un peor pronóstico
a priori
que los pacientes que reciben el tratamiento (ya sea éste el tratamiento
activo o el placebo). Por ello, si se excluyen del análisis a los
pacientes no cumplidores (“análisis por protocolo”), desaparece la
garantía de que los dos grupos sean realmente comparables.
La posición más extendida
aconseja que el análisis se haga “por intención de tratar”, es decir, que
se analicen todos los pacientes que fueron aleatorizados, sin exclusiones,
en el grupo al que fueron asignados inicialmente, independientemente de si
recibieron o no el tratamiento, de que hubiera o no violación del
protocolo, o de que haya fallado o no el seguimiento [9].
Aunque en ocasiones puede parecer ilógico, el análisis por
intención de tratar tiene dos claras ventajas. En primer lugar, esta
estrategia es más coherente con la vida real, donde el no cumplimiento o
las violaciones del protocolo son habituales. Y segundo, y más importante,
el análisis por intención de tratar tiende a preservar la comparabilidad
de los dos grupos.
En lo que resta de este
capítulo retomaremos estos puntos clave dentro del marco de los ensayos
clínicos en pacientes con sepsis. El lector interesado puede ampliar estos
aspectos en la bibliografía recomendada al final de este capítulo.
En los últimos 15 años la
mayoría de los ensayos clínicos en pacientes con sepsis se han apoyado en
las definiciones del American College of Chest Physicians / Society of
Critical Care Medicine (ACCP / SICM) [10]. Estas definiciones han sido
útiles para homogeneizar la nomenclatura y hacer comparables los diversos
estudios; sin embargo, su utilización como criterio único para seleccionar
pacientes en ensayos clínicos es probablemente inadecuada.
En efecto, una primera
característica que debe cumplir la población a estudio es que tenga un
nivel apropiado de gravedad [11]. Como ejemplo hipotético, imaginemos un
ensayo clínico en pacientes con sepsis de amplio espectro, en el que 200
pacientes son asignados al grupo experimental y 200 al grupo control
(figura 2). La mortalidad basal de los pacientes es del 36%, y el
tratamiento reduce la mortalidad en un 25% (Riesgo relativo 0,75). Como se
puede ver en la figura 2, ello supone una reducción absoluta del riesgo
del 9%, lo que significa que hay que tratar a unos 11 pacientes (1/0,09)
para evitar una muerte. Desgraciadamente, la P no alcanza la significación
estadística (P = 0,067), con lo que el ensayo no resulta concluyente.
Figura 2
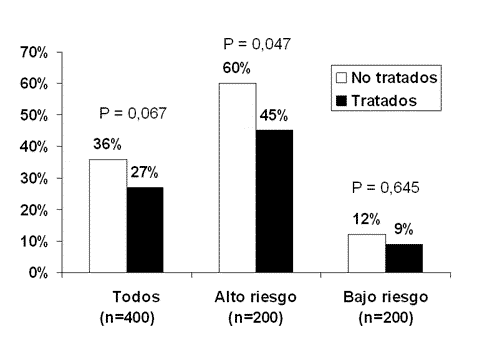
Supongamos ahora que esa
población en estudio está formada por 200 de pacientes de alto riesgo (con
una mortalidad basal del 60%) y 200 pacientes de bajo riesgo (con una
mortalidad basal del 12%). En el grupo de alto riesgo, la mortalidad tras
el tratamiento se reduce al 45% (0,60 x 0,75). La reducción absoluta del
riesgo es ahora del 15%, lo que supone que basta con tratar a 7 personas
para evitar una muerte (1/0,15), y esta asociación es estadísticamente
significativa (P = 0,047). Por lo tanto, el efecto neto de la ampliación
de la muestra con 200 pacientes de bajo riesgo (por ejemplo para hacerla
más “representativa”) ha sido la introducción de “ruido”, con la
consiguiente reducción de la potencia estadística del estudio, y la
conversión del estudio en un ensayo no concluyente.
En el otro extremo, los
pacientes demasiado graves es difícil que se puedan beneficiar de
cualquier tratamiento, por lo que constituyen otra fuente de “ruido” que
puede enmascarar la efectividad del tratamiento. Por ejemplo, en el
estudio PROWESS [12],
tras la inclusión de 720 pacientes, los investigadores decidieron
(apropiadamente) cambiar los criterios de reclutamiento al objeto de
excluir pacientes con alta probabilidad de morir por causas no
directamente relacionadas con la sepsis.
|
Tabla I. Recomendaciones del grupo de trabajo del MRC
sobre ensayos clínicos en pacientes con sepsis [1]
|
| |
| 1.- |
La inclusión de pacientes en el ensayo no debe apoyarse exclusivamente en
las definiciones del American College of Chest Physicians / Society of
Critical Care Medicine. Los criterios de entrada deben basarse en 3
principios: (a) Todos los pacientes deben tener infección; (b) Debe haber
evidencia de un proceso patológico que represente una diana biológicamente
plausible para la intervención; (c) Los pacientes deben caer en una
categoría adecuada de gravedad (generalmente sepsis grave). |
| 2.- |
Los investigadores deben usar un
sistema de puntuación de disfunción orgánica validado. |
| 3.- |
El desenlace primario debe ser en
general la mortalidad, aunque bajo determinadas circunstancias se pueden
considerar como puntos diana primarios la morbilidad. Independientemente
del desenlace elegido, el seguimiento debe prolongarse durante más de 90
días. |
| 4.- |
El tamaño muestral debe basarse en
una estimación realista del tamaño de efecto, basado en el conocimiento de
la población en riesgo. |
| 5.- |
Sólo se deben examinar unos pocos
subgrupos, basados en variables presentes antes de la aleatorización. |
| |
En concordancia con las
consideraciones anteriores, un grupo de expertos reunido por el Medical
Research Council británico para promover unos mejores diseños de los
ensayos clínicos en pacientes con sepsis [1] recomienda que los criterios
de inclusión no se basen exclusivamente en la definición de sepsis del ACCP / SICM, por 2 razones básicas (tabla
I):
a) La
primera es que los pacientes incluidos en los ECA deben tener un riesgo
apreciable de presentar el evento diana, y este alto riesgo no se asegura
con una población heterogénea de enfermos, con pronósticos diversos, que
se agrupan tras la definición de sepsis.
b) La
segunda es que los pacientes deben ser especialmente proclives a
beneficiarse del nuevo tratamiento. Por ejemplo, si se está ensayando un
fármaco que interfiere en la síntesis de la pared bacteriana de los
bacilos gram negativos, no sería lógico seleccionar pacientes con sospecha
de sepsis estafilocóccica. De igual manera, una rotura de cuerdas
tendinosas secundaria a una endocarditis no se va a resolver con un
tratamiento con proteína C activada, por mucho que el paciente cumpla
criterios de sepsis.
|
4. Desenlace
clínico estudiado |
Como regla general, los
ensayos útiles para la toma de decisiones clínicas miden la efectividad
del tratamiento mediante verdaderos desenlaces clínicos (outcomes),
como la mortalidad, la calidad de vida o los días de hospitalización. Los
ensayos basados en desenlaces intermedios como la temperatura [13], o el
número de neutrófilos en el hemograma [14, 15], son importantes para el
investigador interesado en ampliar las fronteras del conocimiento, pero no
son directamente útiles para el clínico.
En ocasiones, un desenlace
intermedio es un claro sustituto del desenlace final. Por ejemplo, el
control de la hipertensión arterial puede considerarse como un sustituto
de la morbilidad cardiovascular. Sin embargo, para que un desenlace
intermedio sea un sustituto válido del desenlace final – y por tanto, sea
potencialmente útil para la toma de decisiones en clínica- se deben
cumplir varios requisitos [16]: (1) debe haber una asociación fuerte y
consistente entre el desenlace sustitutivo y el desenlace clínico; (2)
debe existir clara evidencia de que una mejora en el desenlace sustitutivo (con fármacos de la misma clase y fármacos de otras clases) conlleva una
mejora en el desenlace clínico.
El comité de expertos del
MRC [1] (tabla I) concluyó que el desenlace primario en los ensayos
clínicos en sepsis debe ser, en general, la mortalidad, aunque bajo
determinadas circunstancias se puede considerar como resultado final
primario la morbilidad. Independientemente del desenlace elegido, el
seguimiento debe prolongarse durante más de 90 días.
Cuando la reducción de
eventos es pequeña, la demostración de diferencias estadísticamente
significativas entre los grupos experimental y control requiere tamaños
muestrales excesivamente grandes. Frecuentemente se recurre en estos casos
a desenlaces clínicos agregados (composite
end-points), como la
aparición de muerte o fracaso renal. Desgraciadamente, con demasiada
frecuencia, el aumento de la potencia va acompañado por una mayor
dificultad en la interpretación de los resultados [17].
|
5. Análisis
por intención de tratar |
Como se dijo más arriba, al
objeto de preservar el efecto de la aleatorización (es decir, la
comparabilidad inicial de los dos grupos) en general se aconseja realizar
un análisis por intención de tratar. Como ejemplo, en el estudio PROWESS [12]
se aleatorizaron 1.728 pacientes para recibir el tratamiento experimental
(n = 871) o un placebo (n = 857). Sin embargo, 38 pacientes no recibieron
el tratamiento previsto. En el grupo experimental, 14 pacientes cumplían
algún criterio de exclusión, 4 pacientes estaban moribundos antes de que
se pudiera iniciar la infusión del fármaco y 3 pacientes retiraron el
consentimiento. En el grupo placebo, 15 de los pacientes no cumplían
realmente criterios de inclusión y 2 pacientes estaban moribundos antes de
iniciar la infusión. Uno de los pacientes del grupo experimental que no
recibió el tratamiento se perdió en el seguimiento. Utilizando un criterio
de análisis en la peor de las situaciones posibles para el grupo
experimental (worst case analysis),
los autores consideraron a este paciente como fallecido. A los 28 días
fallecieron 216 pacientes asignados al grupo experimental (incluido el
paciente perdido en el seguimiento) y 268 del grupo control. Los
resultados del análisis por protocolo y por intención de tratar se
presentan en la tabla II. Obsérvese que en el primer caso, el denominador
del riesgo está formado por los pacientes que reciben el tratamiento,
mientras que en el análisis por intención de tratar el denominador lo
constituyen todos los pacientes aleatorizados, hayan recibido o no el
tratamiento.
|
Tabla II. Concordancia en los análisis por intención
de tratar y por protocolo
|
| |
Mortalidad en el estudio PROWESS [12]
|
| |
Grupo
control |
Grupo
experimental |
P |
| Análisis por protocolo |
259/840 (30,8%) |
209/849* (24,7%) |
0,005 |
| Análisis por intención de
tratar |
268/857
(31,3%) |
216/871
(24,8%) |
0,003 |
| |
|
|
|
| * Se ha
excluido la pérdida en el seguimiento |
| |
En el caso del estudio PROWESS, los dos tipos de análisis
ofrecen resultados concordantes. Cuando los dos tipos de análisis ofrecen
resultados discordantes, debe examinarse con detalle la posible asociación
entre las exclusiones post-aleatorización y el grupo asignado y, en último
caso, recurrir a un análisis de sensibilidad para delimitar el impacto de
estas exclusiones [9].
Los ensayos clínicos
aportan información sobre la efectividad
promedio del
tratamiento en los pacientes estudiados. Sin embargo, es razonable pensar
que el efecto del tratamiento no es el mismo en todos los pacientes. Por
ello, resulta intuitivo estratificar los resultados por subgrupos
(hombres/mujeres, diabéticos/no diabéticos, diferentes grupos de edad,
etc.), con el objetivo de clarificar la heterogeneidad de efectos y
facilitar la extrapolación de los resultados a un paciente concreto.
Desgraciadamente, el
análisis de subgrupos es una práctica peligrosa, y los lectores de ensayos
clínicos no siempre somos conscientes del riesgo de falsos positivos (y
falsos negativos) cuando se analizan los resultados en subgrupos [18]. Son
muchas las publicaciones que han advertido de los riesgos de esta práctica
y han dado orientaciones generales para interpretar los resultados de los
análisis de subgrupos. Las más conocidas son, probablemente las de Andrew
Oxman [1] (tabla III):
|
Tabla III. ¿Son reales las diferencias entre
subgrupos?
|
| |
| 1.- |
¿Es la magnitud de las
diferencias clínicamente importante? |
| 2.- |
¿Es la diferencia estadísticamente
significativa? |
| 3.- |
¿La hipótesis es previa o
posterior al análisis? |
| 4.- |
¿El análisis de subgrupos es uno de un
pequeño grupo de hipótesis contrastadas? |
| 5.- |
¿Las diferencias eran intra
o entre estudios? |
| 6.- |
¿Eran las diferencias consistentes entre los
diferentes estudios? |
| 7.- |
¿Existe evidencia indirecta
que apoye la hipotética diferencia? |
| |
Dichas orientaciones
señalan el requisito de que los subgrupos se hayan especificado con
antelación a la realización del ensayo. Si los subgrupos se forman una vez
obtenidos los resultados, la probabilidad de que alguno de los “efectos de
subgrupo” se deba al azar (error de tipo I) aumenta.
En segundo lugar, los
subgrupos deben formarse a partir de las características presentes antes
de la asignación aleatoria. Si comparamos el efecto del tratamiento en
subgrupos según la respuesta al tratamiento (por ejemplo pacientes
respondedores o cumplidores), los grupos no serán ya comparables, se
pierde el efecto de la aleatorización y podemos introducir un sesgo a
favor del tratamiento experimental. En ocasiones, el subgrupo al que
pertenece el paciente no es conocido en el momento de la aleatorización;
por ejemplo, en un ensayo de corticoides en pacientes con sepsis, no es
posible determinar si el paciente tiene o no insuficiencia suprarrenal en
el momento de la aleatorización [19]. Sin embargo, ello no compromete la
validez del estudio si se realiza el oportuno análisis estratificado,
estudiando el efecto separadamente en pacientes con y sin insuficiencia suprarenal relativa.
En tercer lugar, para poder
afirmar que existe un efecto diferencial en un subgrupo de pacientes es
necesario que realmente existan diferencias entre los grupos. Esto, que
parece una perogrullada se olvida frecuentemente cuando el análisis de
subgrupos se basa exclusivamente en la comparación de la P
estadística [20, 21]. En efecto, la P no depende solamente de la magnitud
del efecto. Así, la existencia de una P no significativa en un subgrupo
puede deberse tanto a una falta de efecto en ese subgrupo como a una
insuficiente potencia estadística del estudio. Es por tanto necesario
demostrar que el efecto es realmente distinto en los distintos subgrupos,
es decir, que existe heterogeneidad
estadística.
Como ejemplo (figura 3), en
el estudio PROWESS [12], el uso de proteína C activada redujo el riesgo de
muerte en los pacientes con sepsis de origen pulmonar (RR = 0,75; IC 95%
0,61-0,91; P = 0,0047), mientras que no mostró beneficios en la infección intra-abdominal (RR 0,91; IC 95%: 0,65-1,26; P = 0,5618). Una lectura poco
cuidadosa podría sugerir que el tratamiento experimental es efectivo en
pacientes con sepsis de origen pulmonar e ineficaz en pacientes con sepsis
de origen abdominal. Un examen cuidadoso muestra, sin embargo, un gran
solapamiento entre ambos intervalos de confianza. Y un test de
heterogeneidad muestra una P de 0,3299 (no significativa). Por lo tanto,
no se puede afirmar que exista un efecto de subgrupo.
Figura 3
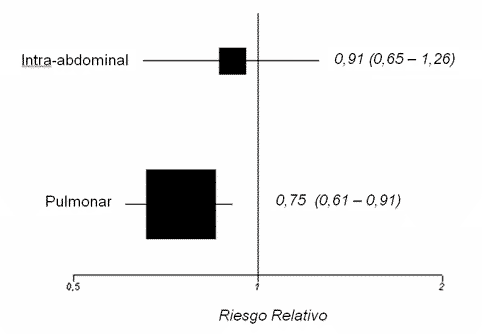
Los pacientes con sepsis
requieren la aplicación de un gran número de medidas terapéuticas y de
sostén, además del tratamiento experimental. Ello hace a los ensayos en
sepsis especialmente propensos a la posibilidad de que se realice un
esfuerzo terapéutico distinto (al margen de la intervención en estudio).
De esta forma, los 2 grupos que inicialmente eran comparables, pueden
hacerse progresivamente divergentes conforme avanza el ensayo debido a que
en un grupo se realiza un mayor esfuerzo terapéutico que en otro. Por
ejemplo, Sprung y col. [22] han comunicado que un 22% de los enfermos
incluidos en un ensayo multicéntrico de sepsis recibieron limitación de
esfuerzo terapéutico, con una mortalidad hospitalaria del 97%. Si esta
limitación de esfuerzo terapéutico no fuera uniforme en los dos grupos, se
estaría introduciendo un sesgo en la estimación del efecto. Este sesgo se
previene habitualmente mediante 2 herramientas: (1) la protocolización
rigurosa de las intervenciones que reciben los pacientes; y (2) el
enmascaramiento (doble ciego), impidiendo que tanto los investigadores
como el propio paciente conozcan el grupo al que han sido asignados.
|
8. Aplicación
individual de los resultados |
El paciente individual rara
vez está adecuadamente representado en un ECA. Ello se debe en parte a
que los ensayos se realizan en pacientes apropiados para evaluar la
eficacia del tratamiento, con criterios de inclusión y exclusión
estrictos, bajo condiciones controladas y con equipos de investigadores
altamente motivados. Todas estas condiciones hacen que los resultados del
ensayo clínico representen el efecto del tratamiento bajo condiciones
óptimas (estudios de “eficacia”).
En la práctica real, bajo condiciones asistenciales no siempre óptimas,
con enfermos con comorbilidad (mayor propensión a los efectos adversos del
tratamiento), los resultados (“efectividad”
real) suelen ser inferiores. Por otro lado, el tratamiento se aplica a un
paciente, con unos determinados valores y preferencias, que es exigible
respetar y que pueden ser incompatibles con la aplicación vertical de la
evidencia.
Por todo ello, no basta
disponer de un ensayo suficientemente válido que demuestre una efectividad
relevante en términos de desenlaces clínicos. Para dar el salto entre los
resultados del ensayo y el paciente individual es preciso estimar la
relación riesgo-beneficio individual y, a ser posible, tomar en cuenta las
preferencias del enfermo (tabla IV).
|
Tabla IV. Aspectos a considerar en la aplicación
individual de los resultados de un ensayo clínico:
|
| |
| 1.- |
Circunstancias locales:
recursos materiales y humanos disponibles |
| 2.- |
Riesgo individual: propensión individual del paciente a
los efectos beneficiosos y adversos del tratamiento |
| 3.- |
Valores del paciente con respecto al tratamiento y a
los desenlaces clínicos del mismo |
| 4.- |
Costes |
| |
Para ilustrar estos
aspectos, partiremos de un escenario hipotético (tabla V).
|
Tabla V. Escenario clínico:
|
| |
| |
Ingresa
en la UCI una paciente de 45 años, con antecedentes de alcoholismo, con un
cuadro de insuficiencia respiratoria por bronconeumonía y shock que no se
remonta con la expansión plasmática. Al ingreso, la paciente está
taquipneica, con 125 lpm, temperatura 38,8º C, TA 60/40 mmHg y una SpO2
del 93% con mascarilla de O2 de alto flujo. La analítica de Urgencias
muestra un APTT de 45”, con un Índice de Quick del 95% y un fibrinógeno de
350 mg/dL. El hemograma (incluido el recuento plaquetario) es normal. Se
inicia tratamiento antibiótico y de sostén y el clínico se plantea la
posibilidad de administrar proteína C activada. |
| |
El estudio PROWESS ha
demostrado que la proteína C activada (PCA) reduce la mortalidad a los 28
días en pacientes similares a la del escenario clínico (24,7% frente a
30,8%, P = 0,005), aunque con tendencia a aumentar la incidencia de
hemorragias graves (3,5% frente a 2%, P = 0,06). Sin embargo, en el ensayo
mencionado se excluyen pacientes con alto riesgo hemorrágico, y nuestra
enferma (mujer, antecedentes de alcoholismo, APTT ligeramente alargado)
presenta un riesgo hemorrágico mayor que el promedio de los pacientes
incluidos en el estudio.
El número de pacientes que
hay que tratar con PCA para evitar un fallecimiento (NNT) se puede
calcular como la inversa de la diferencia absoluta de riesgos entre los
grupos experimental y el control (1/0,061 = 16). El riesgo hemorrágico es
más difícil de calcular; sin embargo, extrapolando la experiencia de la
anticoagulación crónica [23], estimamos que el riesgo de hemorragia grave
es el doble que el del ensayo. En el ensayo, el número necesario para
hacer daño (NNH) es de 67. Por tanto, el NNH ajustado para pacientes
similares a la nuestra es de 34 (67/2)). En otras palabras, cada 34
pacientes como la nuestra tratadas con PCA provocamos 1 hemorragia grave
adicional, pero evitamos 2 fallecimientos, que se hubieran producido sin
PCA. La relación riesgo beneficio es, por tanto, claramente favorable:
aunque las hemorragias producidas por el PCA fueran letales, se evitarían
más fallecimientos administrando PCA que administrando placebo.
Otras veces, sin embargo,
la situación es más comprometida. Por ejemplo, supongamos que, en el
escenario anterior, el riesgo hemorrágico de la paciente es 5 veces
superior a la del ensayo (en la práctica estas situaciones de alto riesgo
hemorrágico entrarían dentro de las contraindicaciones aceptadas del uso
de PCA). Además, debido a las convicciones religiosas de la enferma, para
ella el impacto
de una hemorragia grave que
requiera transfusión es casi tan grave como la muerte. En este caso, puede
ayudar el cálculo del umbral de tratamiento (UA)
[24],
definido como:
Aumento
absoluto de efectos adversos x Impacto de efectos adversos
UA =
-------------------------------------------------------------------------
Número absoluto
de eventos prevenidos x Impacto de eventos prevenidos
En la práctica, el UA no es
más que un refinamiento de la tradicional “relación riesgo/beneficio”.
Cuanto menor sea el umbral de acción, mejor será la relación
riesgo/beneficio. Un UA mayor de 1 indica que el tratamiento tiene más
probabilidades de hacer daño que de beneficiar al paciente y, por lo
tanto, no debería ser administrado.
De acuerdo con la paciente
o su representante, asignamos al fallecimiento un
impacto
de 1 y a la hemorragia grave un impacto
de 0,9.
La reducción absoluta del
riesgo = 1/NNT = 1/16
El aumento absoluto del
riesgo = 1/(NNH/5) = 1/14
El umbral de acción será:
1/14 x 0,9
UA = ------------ = 1,03
1/16 x 1
En este caso, el UA es
superior a 1, con lo que no podemos, de acuerdo con el riesgo individual y
sus preferencias, recomendar al paciente la administración de proteína C
activada.
-
Cohen J, Guyatt G, Bernard GR, Calandra T, Cook D,
Elbourne D et al. New strategies for clinical
trials in patients with sepsis and septic shock. Crit Care Med 2001;
29: 880-886.
-
Cronin L, Cook DJ, Carlet J, Heyland DK, King D,
Lansang MA et al. Corticosteroid treatment for sepsis: a critical
appraisal and meta-analysis of the literature. Crit Care Med 1995;
23: 1430-1439.
-
Ziegler EJ, Fisher CJ, Jr., Sprung CL, Straube RC,
Sadoff JC, Foulke GE et al. Treatment of gram-negative bacteremia and
septic shock with HA-1A human monoclonal antibody against endotoxin. A
randomized, double-blind, placebo-controlled trial. The HA-1A Sepsis
Study Group. N Engl J Med 1991;
324:
429-436.
-
Hotchkiss RS, Karl IE. The pathophysiology and
treatment of sepsis. N Engl J Med 2003; 348:
138-150.
-
Marshall JC. Clinical trials of mediator-directed
therapy in sepsis: ¿what have we learned? Intensive Care Med 2000;
26: S75-S83.
-
Graf J, Doig GS, Cook DJ, Vincent JL, Sibbald WJ.
Randomized, controlled clinical trials in sepsis: has methodological
quality improved over time? Crit Care Med 2002;
30: 461-472.
-
Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical
evidence of bias. Dimensions of methodological quality associated with
estimates of treatment effects in controlled trials. JAMA 1995;
273: 408-412.
-
Egger M, Juni P, Bartlett C, Holenstein F, Sterne J.
How important are comprehensive literature searches and the assessment
of trial quality in systematic reviews? Empirical study. Health Technol
Assess 2003; 7:
1-76.
-
Fergusson D, Aaron SD, Guyatt G, Hébert P.
Postrandomisation exclusions: the intention to trear principle and
excluding patients from analysis. BMJ 2002;
325: 652-654.
-
Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus
WA et al. Definitions for sepsis and organ
failure and guidelines for the use of innovative therapies in sepsis.
The ACCP/SCCM Consensus Conference Committee. American College of Chest
Physicians/Society of Critical Care Medicine. Chest 1992;
101: 1644-1655.
-
Sackett DL. Why randomized controlled trials fail but
needn't: 2. Failure to employ physiological statistics, or the only
formula a clinician-trialist is ever likely to need (or understand!).
CMAJ 2001; 165:
1226-1237.
-
Bernard GR, Vincent JL, Laterre PF, LaRosa SP,
Dhainaut JF, Lopez-Rodriguez A et al. Efficacy and safety of recombinant
human activated protein C for severe sepsis. N Engl J Med 2001;
344: 699-709.
-
Gozzoli V, Treggiari MM, Kleger GR, Roux-Lombard P,
Fathi M, Pichard C et al. Randomized trial of the effect of antipyresis
by metamizol, propacetamol or external cooling on metabolism,
hemodynamics and inflammatory response. Intensive Care Med 2004;
30: 401-407.
-
Weiss M, Voglic S, Harms-Schirra B, Lorenz I, Lasch
B, Dumon K et al. Effects of exogenous recombinant human granulocyte
colony-stimulating factor (filgrastim, rhG-CSF) on neutrophils of
critically ill patients with systemic inflammatory response syndrome
depend on endogenous G-CSF plasma concentrations on admission. Intensive
Care Med 2003; 29:
904-914.
-
Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson
JW. A randomized phase II trial of granulocyte-macrophage
colony-stimulating factor therapy in severe sepsis with respiratory
dysfunction. Am J Respir Crit Care Med 2002;
166: 138-143.
-
Bucher H, Guyatt G, Cook D, Holbrook A, McAlister F.
Surrogate outcomes. In: Guyatt G, Drummond R,
Sande MA, Gilbert DN, Moellering RC, editors.
Users' Guide to the Medical Literature: A Manual for Evidence-Based
Clinical Practice. American Medical Association; 2002.
-
Freemantle N, Calvert M, Wood J, Eastaugh J, Griffin
C. Composite outcomes in randomized trials: greater precision but with
greater uncertainty? JAMA 2003; 289:
2554-2559.
-
Oxman AD, Guyatt GH. A consumer's guide to subgroup
analyses. Ann.Intern.Med 1992;
116: 78-84.
-
Annane D, Sebille V, Charpentier C, Bollaert PE,
Francois B, Korach JM et al. Effect of
treatment with low doses of hydrocortisone and fludrocortisone on
mortality in patients with septic shock. JAMA 2002;
288: 862-871.
-
Matthews JN, Altman DG. Statistics notes. Interaction
2: Compare effect sizes not P values. BMJ 1996;
313: 808.
-
Altman DG, Bland JM. Interaction revisited: the
difference between two estimates. BMJ 2003;
326: 219.
-
Sprung CL, Finch RG, Thijs LG, Glauser MP.
International sepsis trial (INTERSEPT): role and impact of a clinical
evaluation committee. Crit Care Med 1996; 24:
1441-1447.
-
Eckman MH, Jevine HJ, Pauker SG. Making decisions
about antithrombotic therapy in heart disease. Decision analytic and
cost-effectiveness issues. Chest 1995;
108 (Sup 4):
457S-470S.
-
Gross R. Decisions and evidence in medical practice.
Applying evidence-based medicine to clinical practice.
S. Luis: Mosby / Harcourt; 2001.
Jaime Latour Pérez
Hospital Universitario de Elche, Alicante
©REMI,
http://remi.uninet.edu. Febrero 2005.
Palabras clave:
Sepsis grave, Ensayos clínicos, Aleatorización, Ocultación de la secuencia
de aleatorización, Enmascaramiento, Análisis por intención de tratar,
Análisis de subgrupos, Cointervención, Umbral de tratamiento.
Busque en REMI con Google:
Envía tu comentario para su
publicación |
|
|
|



