En 1912 Leber describió un cuadro que denominó "degeneración retiniana con aneurismas miliares múltiples". Reese, en 1956 (24), describió en esta afección las lesiones fundamentales. Según este autor se trataba de telangiectasias retinianas. El cuadro aparecía en población joven y el diagnóstico se realizaba oftalmoscópicamente.
Posteriormente, Duke-Elder llegó a la conclusión que este cuadro era una fase precoz de una enfermedad denominada de Coats. Más adelante, y en la actualidad, todos los autores incluyen la enfermedad de Leber en el contexto de la angiomatosis de Coats.
En 1908 George Coats hizo la primera descripción de la enfermedad que lleva su nombre, aunque la interpretación de la misma la realizó en 1912.
Clínicamente se describen tres formas (7). La mas frecuente es la infantil o enfermedad de Coats propiamente dicha. Suele aparecer en la primera década de la vida entre los 3 y 10 años y generalmente es unilateral. Es un desorden vascular de origen incierto difuso y que afecta a la retina periférica. Otra forma es la denominada juvenil, que se correspondería con la enfermedad de Leber. Es periférica y circunscrita. Se trata de un forma minor. Por ultimo existe una forma infrecuente que es la del adulto y que adopta una disposición yuxtaalveolar.
La forma infantil se presenta oftalmoscópicamente con exudación amarillenta subretiniana o intrarretiniana. Se advierten telangiectasias arteriolares y aneurismas capilares. Es frecuente encontrar alteraciones pigmentarias, cristales de colesterol y desprendimiento retiniano exudativo. La exudación retiniana es progresiva. Si la retina se desprende y se coloca detrás del cristalino se aprecia una leucocoria (Fig. 1) semejante a la del retinoblastoma. Cuando las lesiones evolucionan hasta estadios extremos el cuadro clínico se complica con un glaucoma.
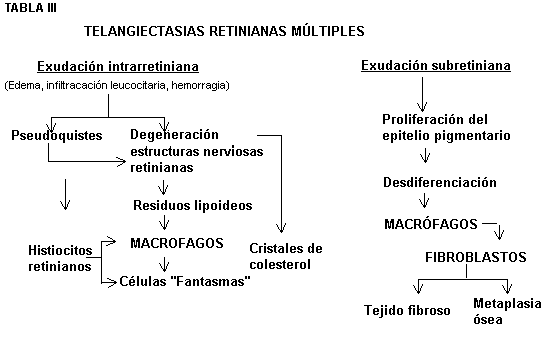
Anatomopatológicamente las lesiones son muy variadas tanto en la retina como fuera de ella (7,33). La retina se encuentra casi siempre desprendida (Fig. 2) y se aprecia un engrosamiento importante de la misma debido a la presencia de edema y exudados. Estos son densos, amorfos, hialinos, eosinófilos PAS+ y ricos en glicoproteínas. Esto origina una gran desorganización en las capas retinianas con formación de quistes y degeneración de la estructuras nerviosas retinianas. Ello lleva consigo la presencia de residuos lipoideos, acompañados de cristales de colesterol (Fig. 3). Ese material residual es fagocitado por histiocitos retinianos apareciendo las células multinucleadas denominadas "células fantasmas" (Fig. 4). En el espacio subretiniano también se produce exudación. Esta estimula al epitelio pigmentario cuando evoluciona la enfermedad el cual prolifera y se desdiferencia dando lugar a la formación de fibroblastos que son los responsables de la aparición del tejido fibroso. No es infrecuente la presencia de tejido óseo metaplásico (7,34).
Las lesiones mas importantes son las vasculares. Los vasos
arteriales presentan sus paredes engrosadas a expensas de un
material PAS+. La imagen morfológica es semejante a la de una
necrosis fibrinoide. En ocasiones algunos de estos vasos se
encuentran trombosados con material amorfo intraluminal. Los
vasos de pared fina por el contrario se suelen encontrar
dilatados y su pared también se tiñe con el PAS constituyendo
las conocidas Telangiectasias (Fig. 5), lesión característica de la
enfermedad. Estas alteraciones vasculares y sus lesiones
acompañantes (edema, lipofagia e infiltración leucocitaria
perivascular) son sobre las cuales descansan las hipótesis
patogenéticas. Se sabe hoy que el carácter primario de las
hemorragias tal como lo desarrollo Coats es falso. Numerosos
autores han reconocido en los últimos años que son las
alteraciones vasculares la causa primera de la enfermedad. Manchostt y Brujint (17) han sido los
primeros en afirmar que son las telangiectasias las que ponen en
marcha todo el mecanismo fisiopatólogico de la enfermedad. La
hipótesis patogénica se expone en la tabla III.
¿Por qué se producen las telangiectasias? Existen múltiples teorías. La primera publicada, se realizó en 1934 por Junius quien decía que en las arteriolas se producían fenómenos físico-químicos que darían lugar a cambios en los tejidos por ella nutridos. Wise (42) responsabiliza a la hipoxia local de los cambios vasculares. Últimamente se cree que el factor patogénico mas importante sería una alteración de la permeabilidad capilar. Por otro lado teniendo en cuenta que las lesiones de la enfermedad de Coats se asemejan a vasculitis nodular por hipersensibilidad, es por lo que se ha pensado que esta afección surge como consecuencia de una vasculitis alérgica retiniana.
![]()
![]()
![]()