|
| |
|
Revista
Electrónica de Medicina Intensiva
Artículo nº C20. Vol 4 nº 12, diciembre 2004.
Autores: Abelardo García de Lorenzo Mateos, Manuel Quintana Díaz
|
[ Anterior ] [ Arriba ] [ Siguiente ]
|
-
Inflamación y coagulación
-
Trombosis y hemorragia
-
Anemización. Papel de las transfusiones y la eritropoyetina
-
Conclusiones
-
Bibliografía
La sepsis tiene una fisiopatología
compleja y no totalmente elucidada, en la que están involucradas la
respuesta inflamatoria, las alteraciones de la hemostasia y la disfunción
endotelial [1].
Un entendimiento fisiopatológico más global
de esta entidad clínica sería aquel que considera una respuesta del
huésped que incluye la activación de la respuesta inflamatoria, la
activación de la cascada de la coagulación y la inhibición de los
mecanismos fibrinolíticos. Esta respuesta incluye una compleja interacción
entre la respuesta inflamatoria y la de la coagulación que conduce a una
disfunción endotelial generalizada.
Parece suficientemente demostrada la
prevalencia de cifras elevadas de los marcadores pro-inflamatorios en los
pacientes de riesgo ingresados en las UCIs, siendo éstos útiles tanto en
la detección precoz de procesos inflamatorios e infecciosos como en la
predicción del riesgo de complicaciones y/o mortalidad [2].
El endotelio vascular, en
condiciones normales, regula la formación del coágulo y su lisis. Sus
funciones pueden estar alteradas por la exposición a agentes infecciosos o
a mediadores inflamatorios como la endotoxina bacteriana, los componentes
del complemento, la interleucina (IL) IL-1, el factor de necrosis tumoral
(FNT) o las proteasas granulocitarias [3].
El endotelio lesionado o
estimulado manifiesta funciones procoagulantes: puede expresar factor
tisular (FT), disminuir la trombomodulina, liberar el inhibidor del
activador del plasminógeno (PAI), liberar factor activador de las
plaquetas, endotelina y factor von Willebrand, así como disminuir la
síntesis de óxido nítrico [4]. La exposición del subendotelio a la sangre
produce adhesión y agregación plaquetaria, así como la puesta en marcha de
los mecanismos de la coagulación. La endotoxina incrementa la adhesión de
granulocitos y monocitos al endotelio capilar y causa una lesión
endotelial difusa, mediada por IL-1 y FNT [5] .
El FT es una glucoproteína
de membrana perteneciente a la familia de los receptores de las citocinas
(como los del interferon alfa y gamma), presente en la membrana plasmática
de diversos tipos celulares (adventicia de vasos sanguíneos, piel, tejido
nervioso, glomérulo renal, líquido amniótico...), que activa el inicio
de la cascada de las serinproteasas del proceso de la coagulación [5].
El FT no se expresa
normalmente en la pared vascular, a excepción de una expresión moderada en
la adventicia [6]; sin embargo, tanto en los monocitos como en las propias
células endoteliales puede ser inducida por la exposición a endotoxinas
bacterianas, ésteres de forbol, lectinas, factores de crecimiento y
mediadores celulares de la respuesta inmunológica (linfocitos T) [7]; no
obstante, para ser inductor de la coagulación precisa de una remodelación
de la bicapa lipídica de la membrana plasmática [8], y es que en presencia
de fosfolípidos aumenta espectacularmente su afinidad por el FVII
activado.
Sin embargo, con la
excepción de la meningococemia grave [9], se ha comprobado que es difícil
demostrar la expresión “in vivo” de FT en los monocitos de pacientes
sépticos o de animales de experimentación expuestos sistemáticamente a
microorganismos.
En resumen, podríamos
afirmar que se han acumulado pruebas indicativas de que en la relación
entre inflamación y activación de la coagulación podrían estar implicados
mecanismos más complejos [10]. Las células endoteliales responden a las
citocinas expresadas y liberadas por los leucocitos activados, pero ellas
a su vez también pueden liberar citocinas (Figura 1). Además, las células endoteliales son capaces de expresar moléculas de adhesión y factores de
crecimiento, que no sólo promueven aún más la respuesta inflamatoria, sino
que también pueden afectar a la respuesta de la coagulación.
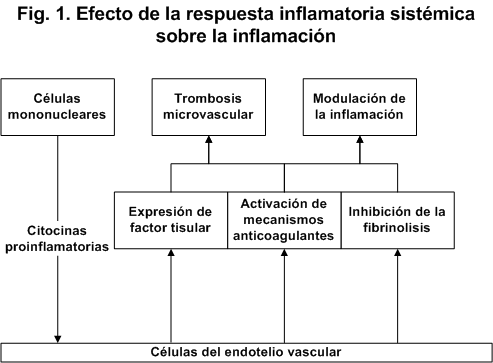
Es interesante señalar que
ambos sistemas, inflamación y coagulación, interactúan, pudiendo la
coagulación modular la respuesta inflamatoria [11]. Esta compleja
interrelación entre los dos sistemas es importante en muchos
estados de enfermedad, entre ellos el síndrome de respuesta inflamatoria
sistémica, que pueden dar lugar a disfunción orgánica y mortalidad en la
sepsis.
Tras la agresión
se desencadena una serie de respuestas
orgánicas programadas que tienden a limitar el cuadro inflamatorio. Los
trastornos del sistema de la coagulación son consecuencia de la causa
desencadenante, y sea por la gravedad o duración de la agresión, o por las
específicas condiciones del paciente, no se limita al punto lesionado y da
lugar a unos cuadros clínicos que son consecuencia del desequilibrio de la
hemostasia normal y que tienen su máxima expresión en forma de trombosis
y/o hemorragias [12].
Las vías patogénicas
asociadas a la coagulopatía en la sepsis son [11, 12]:
-
Generación de trombina
mediada por FT, como respuesta inflamatoria, que conlleva formación y
depósitos en la microcirculación.
-
Vías anticoagulantes disfuncionales,
por inhibición progresiva de la anticoagulación endógena por consumo
progresivo.
-
Bloqueo e inhibición de la fibrinolisis,
inicialmente eficaz
-
Disfunción plaquetaria por diversos
mecanismos.
Durante la infección grave, la
activación esta mediada por el FT, que al expresarse en la superficie
celular de las citocinas interactúa con el factor VII, tanto en su forma
cimógena (FVII) como activada (FVIIa), poniendo en marcha la cascada de
la coagulación, que conlleva la conversión de protrombina en trombina.
Como consecuencia de esta generación de
trombina se activan las vías anticoagulantes fisiológicas, que operando
desde el endotelio protegen la microvascularización del estrés hemostático
e inflamatorio. La generación de trombina esta limitada por:
-
La antitrombina (AT), que al unirse bloquea su acción proteolítica. Durante
los procesos sépticos las concentraciones de AT son bajas debido al
consumo, alteración de la síntesis y a la degradación por la elastasa
procedente de los neutrófilos activados [13],
influyendo en este descenso la reducción de la disponibilidad de
glucosaminoglucanos a nivel de la superficie endotelial perturbada, ya
que actúan de manera similar a la heparina, es decir como cofactor
fisiológico de la AT [14].
-
El
sistema de la proteína C, que es trombina dependiente, formando un
complejo al unirse a la trombomodulina (molécula asociada a la membrana
de la célula endotelial), y que actúa, una vez activada y potenciada por
el cofactor S, sobre la cascada de la coagulación decelerando al
inactivar a los FVa y FVIIIa mediante escisión proteolítica [15].
Sin embargo, en la sepsis, además de niveles ya bajos, las citocinas
proinflamatorias, TNF-alfa e IL-1, regulan a la baja la expresión de
trombomodulina, disminuyendo por tanto su activación [16].
-
El
inhibidor de la vía del FT (TFPI), que existe asociado a las células
endoteliales o unido a las lipoproteínas en el plasma. Es factor Xa
dependiente y bloquea la acción del FT-VIIa al formar un complejo
cuaternario (FT-VIIa-TFPI-Xa). Aunque su importancia no se ha conseguido
demostrar en estudios clínicos, los efectos beneficiosos, demostrados en
conejos y babuinos, e intuidos en voluntarios sanos [17],
pueden ser debidos a su efecto atenuante de la generación de IL-6 y a su
capacidad para unirse a la endotoxina e interferir con su interacción
con las CD14, es decir en la producción de un bloqueo de la vía
procoagulante desencadenada por la endotoxina [18].
-
La
prostaciclina, que es trombina dependiente y que disminuye la agregación
plaquetaria por disminución de la unión fibrinógeno-plaquetas al
producir un descenso en la expresión den la superficie plaquetar de la
glicoproteina IIb/IIIa.
En la modulación de la activación de la
coagulación como respuesta a la agresión, además de los anticoagulantes
fisiológicos, interviene el sistema fibrinolítico, que es un elemento
clave de la patogenia del depósito de fibrina durante la inflamación
grave.
Los principales activadores e
inhibidores de la fibrinolisis son sintetizados y almacenados por las
células endoteliales. Aunque la respuesta inicial en la bacteriemia y en
la endotoxemia es un aumento de la activación fibrinolítica, mediada por
la liberación casi inmediata de activadores del plasminógeno tisular,
t-PA, que se acompaña de una elevación de los complejos plasmina-α-2antiplasmina
en plasma, su duración es corta y rápidamente es suprimida por un aumento
mantenido del principal inhibidor de la fibrinolisis, el inhibidor del
activador del plasminógeno 1 (PAI-1), al estar agotada la liberación de
t-PA [19]. Es decir, que se
produce una inhibición de la fibrinolisis, que es inicialmente eficaz [12].
Por último, como hemos
señalado, dentro de las vías patogénicas asociadas a la coagulopatía en la
sepsis está la disfunción plaquetaria, tanto en recuento total como en
función. Así, en el paciente crítico de origen séptico, se objetivan
variaciones en los recuentos plaquetarios, que presentan un patrón
bifásico que difiere en los supervivientes y en los no supervivientes,
encontrando que la trombocitopenia tardía es más predictiva de muerte que
la precoz [20].
Las plaquetas pueden estar
afectadas además de por el propio proceso séptico (que asocia fallo renal
y hepático) por la propia terapéutica, sangrado concomitante, etc., y así se
produce una variabilidad de la adhesividad y
agregabilidad por cambios en sus receptores de membrana por degranulación
(alfa y beta) secundaria a PDF tardíos (D y E) [12]. El descenso en el
número de plaquetas se produce tanto por hemofagocitosis (destrucción de
células hematopoyéticas por monocitos y macrófagos) como por destrucción
en el espacio microvascular y su secuestro en diversos órganos (hígado,
pulmón, intestino), a lo que habría que sumar el que resulta de la
formación de agregados plaquetarios por aumento en la superficie
plaquetaria de moléculas de adhesión, que facilitan su unión al
fibrinógeno y a la trombospondina [12].
|
3. Anemización.
Papel de las transfusiones y la eritropoyetina |
La anemia
asociada a enfermedad crítica se define como la anemia del paciente
críticamente enfermo que no puede ser explicada por otras causas y que se
caracteriza por una respuesta inadecuada a la eritropoyetina endógena en
relación al grado de déficit de hemoglobina (Hb) presente [21]. Se
considera el hallazgo de laboratorio más frecuente en las UCIs y se ha
referido que a los tres días de su ingreso el 95% de los pacientes
presentan cifras bajas de hemoglobina (menos de 12 y 13 g/dl de
hemoglobina para mujeres y hombres, respectivamente). Si pensamos en el
paciente de UCI, cifras de 10 g/dl de hemoglobina o de hematocrito de 30 %
no suelen “hacer saltar las alarmas” aunque Hebert y col. [22], empleando
un criterio restrictivo, rebaja a 7 g/dl la cifra mínima de Hb o Hb óptima
suficiente para que un paciente (sin compromiso coronario e ingresado en
UCI) sea considerado como candidato a transfusión.
La anemia del
paciente crítico es hematológicamente similar a la anemia de la enfermedad
crónica en lo referente a que el paciente presenta disminución en las
concentraciones séricas de hierro y en la capacidad total de ligazón al
hierro, una baja razón hierro sérico / capacidad total de ligazón al
hierro, y concentraciones normales o hasta elevadas de ferritina sérica. A
pesar de la presencia de anemia, las concentraciones séricas de
eritropoyetina están solo discretamente elevadas en el momento de su
ingreso, sin objetivarse aumentos significativos en el recuento de
reticulocitos circulantes.
La anemia del
paciente crítico no está completamente entendida, pero el proceso puede
ser parcialmente explicado por el efecto de las citocinas sobre la
eritropoyetina. La maduración normal de las células eritroides en hematíes
es regulada a través de la unión y activación de los receptores de la
eritropoyetina por su hormona. La producción renal de la hormona
eritropoyetina es estimulada en condiciones de hipoxia y suprimida como
respuesta a las citocinas inflamatorias, lo que subsecuentemente afecta a
la expresión de los genes de la eritropoyetina (figura 5). Las citocinas
inflamatorias se asocian con una caída de la respuesta eritropoyética en
los pacientes críticos. Además, las citocinas parecen inhibir la
producción de hematíes directamente en la médula ósea y estimular las
proteínas ligadas al hierro que secuestran a éste y limitan la producción
de hematíes en los pacientes críticos [23]. La anemia del paciente crítico
puede tener efectos deletéreos sobre las variables hemodinámicas y sobre
el transporte de oxígeno.
Durante su
estancia en la UCI un paciente tipo pierde más de 700 ml de sangre solo en
pruebas de laboratorio [24]. Por otra parte se ha descrito que en un
paciente crítico, sin hemorragia activa demostrable, la concentración de
hemoglobina disminuye en más de 0,5 g/dl por día durante los primeros días de
estancia. A partir del tercer día la hemoglobina puede mantenerse
relativamente constante en los pacientes no sépticos, pero continúa
disminuyendo en los pacientes sépticos así como en los pacientes con
APACHE II elevado o con presencia de disfunción de órganos de origen séptico (Tabla I).
Estas cifras no se relacionan con el balance hídrico, aunque si presentan
una correlación estadísticamente significativa con el volumen de extracción,
que es mayor en los pacientes sépticos [25]. Por otra parte, la
hemorragia activa -como causa de anemización- no es infrecuente en UCI.
Puede ser secundaria a sangrado gastrointestinal, procedimientos
quirúrgicos, drenajes, etc. Se ha estimado que el 30 % de los pacientes
críticos presentan hemorragia activa, requiriendo transfusión el 35 % de
ellos.
|
Tabla I: Patrón hemático en el
paciente crítico
|
| |
| Hb 10-11 gr/dl,
con: elevación
de ferritina,
¯
Fe ,
¯¯ eritropoyetina y respuesta disminuida de los reticulocitos |
| |
|
Trombocitopenia (¯
30%) |
| |
|
¯
Hb de 0,5 gr/dl los tres primeros días, después estabilización |
| |
| En sepsis,
aún en ausencia de hemólisis o sangrado activo,
¯ Hb de 0,8 gr/dl los tres primeros días, después
¯ continuado de 0,3 gr/dl diarios |
|
|
En lo que
respecta al manejo de la anemia en UCI debemos considerar que es una labor
compleja que afecta a un sumatorio de opciones terapéuticas, entre las que
destacan el hierro, la sangre alogénica, los agentes eritropoyéticos y el
evitar las pérdidas hemáticas (rFVIIa ...).
El objetivo
fundamental del manejo de la anemia es el de mejorar la oxigenación
tisular. Las transfusiones sanguíneas pueden mejorar la capacidad de
transporte de oxígeno en los pacientes críticos, aunque la indicación
indiscriminada de transfusiones con ese objetivo no es efectiva en todos
los pacientes. En 13 ensayos clínicos se valoraron las modificaciones en la
oxigenación tras la transfusión: el aporte de oxígeno aumentó en todos los
casos, pero el consumo de oxígeno solo aumentó en 5 casos (¿dintel de
anaerobiosis?).
En lo que respecta al manejo de la
anemia, las actuales recomendaciones son:
-
Las transfusiones de sangre
alogénica son todavía la forma mas efectiva de aumentar rápidamente
los niveles de hemoglobina, pero las transfusiones indiscriminadas deben
ser evitadas. En el especial contexto del paciente séptico, se debe
indicar transfusión cuando la Hb es menor de 7,0 g/dl y el objetivo
terapéutico debe de ser conseguir unos niveles de Hb entre 7,0 y 9,0
g/dl. No podemos olvidar el lúcido artículo de Corwin y col. en el que
puntualizó diferentes problemas ligados a la mala indicación de la
transfusión en el contexto del paciente críticamente enfermo (Tabla II)
[26].
Pero además debemos recordar que no solo tenemos el problema de las
infecciones, sino también el de los errores e incompatibilidades en la
administración [27] (Tablas III y IV). En los trabajos de Vincent y col.
[28] y de Taylor y col. [29] se refiere la existencia de correlación
positiva entre el número de unidades transfundidas, la presencia de fracaso multiorgánico
y el aumento de la mortalidad. En esta línea cabe
destacar el reciente trabajo de Shorr y col., que encuentra aumento de la
neumonía asociada a ventilación mecánica en el grupo de pacientes
críticos que recibe transfusión (valor independiente y
dosis-respuesta) [30].
La demanda de hierro se incrementa
durante la eritropoyesis, y el paciente anémico generalmente requiere
200 mg/día de Fe exógeno elemental. El tratamiento con hierro parenteral
(1000 mg de Fe dextrano), aún mas efectivo que el aporte por vía oral,
tiene una efectividad limitada en los pacientes críticos debiendo
monitorizarse estrechamente su aporte aún a pesar de emplearse las
nuevas formas galénicas de hierro; esto debe de ser especialmente
considerado en los pacientes sépticos y en los que presentan disfunción
multiorgánica.
Se han documentado aumentos en los niveles de
hemoglobina (componente esencial del aporte de oxígeno) y en el recuento
de reticulocitos junto con disminución del número de transfusiones
requeridas como respuesta a la terapia con epoietina alfa [31]. Las dosis empleadas en los diferentes estudios -con
o sin Fe y/o ácido fólico- oscilan entre 600 y 300 U/kg 3 veces por
semana o 40.000 U/semana. Actualmente no existe una indicación clara de
la eritropoyetina en la anemia de origen séptico.
No aportar plasma fresco congelado para corregir
valores de laboratorio alterados en los parámetros de coagulación si no
existe hemorragia o no se plantean procedimientos invasivos.
Aportar plaquetas cuando el recuento sea menor de
5.000/mm3, independientemente de la presencia de sangrado.
Transfundir plaquetas si el recuento está entre 5.000 y 30.000 y existe
riesgo significativo de sangrado [32].
Por otra parte, no podemos olvidar los agentes
transportadores de oxígeno, o substitutos sanguíneos que no tienen los
problemas ligados a la infecciones o al almacenamiento de sangre; aunque
ninguno está aceptado para uso clínico, existen buenas perspectivas con
los perfluorocarbonos y con las soluciones de hemoglobina soluble.
Finalmente, debemos efectuar una serie de recomendaciones:
Emplear guías clínicas para
indicar la transfusión.
No usar, de rutina, determinaciones de hierro o ferritina.
No usar, de rutina, plasma fresco congelado.
No usar, de rutina, antitrombina.
Aportar Fe intravenoso cuando se considere indicado.
Cuando exista indicación clínica, aportar 40.000 U/semana
de EPO alfa por vía subcutánea, no antes del tercer día de estancia en
UCI.
Emplear EPO alfa junto con Fe.
Cada vez está más clara la importancia de la
interacción entre coagulación e inflamación durante la infección grave y
la sepsis, que en su forma más extrema se manifiestan por CID y fracaso
multiorgánico.
La sepsis grave se caracteriza por la respuesta
sistémica del huésped a una infección, que incluye la activación de la
inflamación, la coagulación y la alteración de la fibrinolisis endógena,
que conduce a lesión endotelial, hipoperfusión, disfunción multiorgánica y
muerte.
Así que tras el
estímulo infeccioso que ha desencadenado la activación de la cascada de la
coagulación con generación de trombina, se han puesto en marcha unos
mecanismos reguladores y limitadores que son llevados a cabo por los
anticoagulantes fisiológicos, pero si el estímulo es importante y
persistente, se ven “desbordados”, y se produciría una generación
descontrolada de trombina, que inicialmente estaría regulada por el
sistema fibrinolítico, que es inicialmente eficaz, pero posteriormente, al
persistir el estímulo y por tanto la activación de la coagulación, se
inhibiría por bloqueo. A esta disfunción en la coagulación se sumaría la
alteración plaquetaria tanto en número (trombocitopenia), como en
función, objetivándose variabilidad en su adhesión y agregabilidad.
La activación masiva de la coagulación que se produce
como consecuencia de una agresión por agente infeccioso y se amplifica por
la respuesta inflamatoria, conlleva en mayor o menor grado la oclusión trombótica de la microcirculación por depósitos de fibrina, generación de
enzimas proteolíticas de la coagulación, que pueden tener efectos
proinflamatorios, y consumo de plaquetas y factores de la coagulación, que
son los responsables de desequilibrios en los dominios funcionales del
sistema hemostático, dando lugar a expresiones clínicas de trombosis y/o
hemorragia.
La anemia asociada a la enfermedad crítica se considera
el hallazgo de laboratorio más frecuente en las UCIs, y se considera hematológicamente
similar a la de la enfermedad crónica, siendo este proceso parcialmente
explicado por el efecto de las citocinas proinflamatorias sobre la
eritropoyetina (asocian una caída de la respuesta eritropoyética).
Además del efecto deletéreo que sobre las variables
hemodinámicas y sobre el transporte de oxígeno tiene la anemia, éste se
agrava con el sangrado activo (gastrointestinal, procedimientos
quirúrgicos, drenajes...), que afecta al 30% de los pacientes críticos y
que obliga a transfundir al 35% de ellos.
En lo que respecta al manejo de la anemia en UCI
debemos considerar que existe un sumatorio de opciones terapéuticas
encaminadas tanto a compensar las pérdidas (aporte de hierro, de sangre
alogénica, de agentes eritropoyéticos) como a evitarlas (minimizar
extracciones, rFVIIa...).
-
Huete
T. Drotecogina alfa (activada). Proteina C activada humana de origen
recombinante en el tratamiento de la sepsis grave. En Net, editor.
Avances y tecnología en Medicina Intensiva. Barcelona: Masson, 2004.
-
Gabay, Kushner I. Acute phase proteins and other systemic responses to
inflammation. N Eng J Med 1999; 86: 501-504.
-
Ruf W, Edgington TS. Structural biology of tissue factor, the initiator
of thrombogenesis in vivo. FASEB J 1994; 8: 385-390.
-
Jaffe EA. Endothelial cell structure and function. En Hoffman R, Benz EJ,
Shattil SJ, Furie B, Cohen HJ, editors. Hematology. Basic principles and
practice. New York: Churchill Livingstone, 1991.
-
Rapaport Sl, Rao VML. The tissue factor pathway: how it has become a
prima ballerina. Thromb Haemostas 1995; 74: 180-184.
-
Velasco
F, López-Pedrera Ch, Dobado-Berrios PM, Torres A. Activación de la vía
extrínseca en la coagulación intravascular diseminada: fisiopatología y
control terapéutico. Rev Iberoamer Tromb Hemost 2000; 13 (Supl
1): 99-107.
-
Edginton TS, Mackman N, Brand K, Ruf W. The structural biology of
expression and function of tissue factor. Thromb Haemost 1991; 66: 67-79.
-
Rapaport SL, Rao VML. Initiation and regulation of tissue
factor-dependent blood coagulation. Arterioscler Thromb 1992;
12: 1111-1121.
-
Osterud B, Flaegstad T. Increased tissue thromboplastin activity in
monocytes of patients with meningococcal infection: Related to an
unfavourable prognosis. Thromb Haemost 1983; 49: 5-7.
-
Marshall JC. Inflamation, coagulopathy, and the pathogenesis of multiple
organ dysfunction syndrome. Crit care Med 2001; 29: S99-S106.
-
Levi M, van der Poll T. The central role of the endothelium in the
crosstalk between coagulation and inflammation in sepsis. En Advances in
sepsis 2004; 3: 91-97.
-
Quintana M, Cabestrero D, García de lorenzo A. Coagulación y hemorragia
en el paciente crítico: patrón, pruebas diagnósticas y etiología. Med
Intensiva 2003; 27: 605-614.
-
Levi M, ten cate H, van der Oll T. Disseminated intravascular
coagulation: state of the art. Thromb Haemost 1999; 82: 695-705.
-
Bourin MC, Lindahl U. Glycosaminoglycans and the regulation of blood
coagulation. Biochem J 1993; 289: 313-330.
-
Levi M, de Jonge E, van der Poll T. Rationale for restoration of
physiological anticoagulant pathways in patients with sepsis and
disseminated intravascular coagulation. Crit Care Med 2001; 29: S90-S94.
-
Faust SN, Levin M, Harrison OB. Dysfunction of endothelial protein C
activation in severe meningococcal sepsis. N Engl J Med 2001;
345: 408-416.
-
Novotny WF, Brown SG, Miletich JP et al. Plasma antigen levels of the
lipoprotein-associated coagulation inhibitor in patient samples. Blood
1991; 78: 387-393.
-
de Jonge E, Dekkers PE, Creasey AA et al. Tissue factor pathway
inhibitor (TFPI) dose-dependently inhibits coagulation activation
without influencing the fibrinolytic and cytokine response during human
endotoxemia. Blood 2000; 95: 1124-1129.
-
van der Poll T, de Jonge E, Levi M. Regulatory role of cytokines in
disseminated intravascular coagulation . Semin Thromb Hemost 2001;
27: 639-651.
-
Akca S,
Haji-Michael P, de mendonca A et al. Time course of
platelet counts in ritically ill patients. Crit Care Med 2002; 30: 753-756.
-
Rudis MI, Jacobi J, Hassan E, Dasta JF.
Managing anemia in the critically ill patient.
Pharmacotherapy 2004; 24:
229-247.
-
Herbert PC, Wells G, Blajchman MA et al. A multicenter,
randomized, controlled clinical trial of transfusion requirements in
critical care. NEJM 1999; 340: 409-417.
-
Corwin HL,Krantz SB. Anemia in the critically ill:
“acute” anemia of chronic disease. Crit Care Med 2000; 28: 3098-3099.
-
Smoller BR, Kruskall MS. Phlebotomy for diagnostic
laboratory test in adults: pattern of use and effect on transfusion
requirements. NEJM 1986; 314: 1233-1235.
-
Nguyen V, Bota PB, Melot C, Vincent JL. Time course of
hemoglobin concentrations in nonbleeding intensive care unit patients.
Crit Care Med 2003; 31: 406-410.
-
Corwin HL, Parsonnet KC, Gettinger A. RBC transfusion in
the ICU. Is there a reason ? Chest 1995; 108: 767-771.
-
Goodnough LT. Risks of blood transfusion. Crit Care Med
2003; 31: S678-S686.
-
Vincent JL, Baron JF, Reinhart K et al. Anemia and blood
transfusion in critically ill patients. JAMA 2002; 288: 1499-1507.
-
Taylor RW, Manganaro L, O´Brien J et al.
Impact of allogenic packed red cell transfusion on
nosocomial infection rates in the critically ill patient. Crit Care Med
2002; 30: 2249-2254.
-
Shorr AF, Duh M-S, Kelly KM et al. Red blood cell
transfusion and ventilator-associated pneumonia: A potential link ?.
Crit Care Med 2004; 32: 666-674.
-
Corwin HL, Gettinger A, Pearl RG et al. Efficacy of
recombinant human erythropoietin in critically ill patients: a
randomized controlled trial. JAMA 2002; 288:
2827-2835.
-
Surviving Sepsis Campaign Guidelines for Management
of Severe Sepsis and the Septic Shock. Society Critical Care Medicine.
Revised June 2004.
Abelardo García de Lorenzo y Mateos, Hospital La Paz, Madrid
Manuel Quintana Díaz, Hospital Virgen del Prado, Talavera, Toledo
©REMI,
http://remi.uninet.edu. Diciembre 2004.
Palabras clave:
Sepsis, Anemia del enfermo crítico, Eritropoyetina, Transfusión,
Hemorragia, Inflamación, Coagulación intravascular diseminada, Trombosis.
Busque en REMI con Google:
Envía tu comentario para su
publicación |
|
|
|



