
3. ETIOPATOGENIA
Las lesiones de PTT-SHU involucran a las arteriolas terminales y capilares,
formando microtrombos compuestos principalmente de plaquetas y fibrina
en menor proporción, al contrario de lo que ocurre en la coagulación
intravascular diseminada (CID) 1,
2. A menudo
se asocian a microaneurismas y depósitos hialinos en el subendotelio
vascular, pero carecen de necrosis fibrinoide y de infiltración
perivascular propia de las vasculitis 6.
La trombosis microvascular, característica de PTT-SHU produce disfunción
isquémica de los órganos, siendo los más frecuentemente
comprometidos: cerebro, riñón, vísceras abdominales
y corazón, aunque puede afectarse todo el organismo (incluyendo
ojos y pulmón).
Los hematíes se deterioran como consecuencia de la interacción con los microtrombos y la red de fibrina de los pequeños vasos (hemólisis "microangiopática" y microangiopatía trombótica). Estos eventos generan microesferocitos y esquistocitos, con deformabilidad limitada que se destruyen con rapidez en el bazo y la microcirculación. Las plaquetas se consumen en los trombos intravasculares o experimentan daño en la circulación, siendo eliminados por el sistema reticuloendotelial 7. La oclusión vascular generalizada (cerebro, riñón, abdomen y corazón) determina el fallo multiorgánico (FMO).
Aunque parece indudable que las manifestaciones propias de la PTT-SHU derivan de lesiones trombóticas microvasculares, los mecanismos patogénicos son heterogéneos. La etiología en la mayor parte de los casos es desconocida, caracterizando la forma idiopática de PTT-SHU. No obstante, se han descrito cuadros de PTT-SHU secundarios o asociados a distintos factores ambientales (infecciones bacterianas y víricas, incluyendo VIH 8), uso de medicamentos y vacunas, diferentes enfermedades autoinmunes y oncológicas, así como la aparición o agravamiento durante embarazo y puerperio (Tabla 1). Sin embargo en la mayoría de estos pacientes no está clara la vinculación entre el presunto factor etiológico y el mecanismo fisiopatogénico subyacente.
Entre los mecanismos y factores actualmente implicados en la patogénesis se encuentran:
3.2. FACTORES INMUNOLÓGICOS, dada la presentación de PTT-SHU en pacientes con enfermedades reumáticas (lupus eritematoso sistémico, artritis reumatoide) 15, 16. La coexistencia de vasculitis y microangiopatía trombótica, induce a pensar que la vasculitis autoinmune podría provocar el daño endotelial desencadenante de la PTT. Sin embargo tal asociación podría ser casual, debido a la mayor incidencia de ambas patologías en mujeres entre 30-40 años 17. En contra del origen inmune, varios estudios demuestran la imposibilidad de transferir la enfermedad infundiendo plasma de pacientes enfermos a individuos sanos 18, al contrario de lo que ocurre en la púrpura trombocitopénica idiopática (PTI) 19.
3.3. DAÑO ENDOTELIAL Y ALTERACIONES EN LA AGREGACIÓN PLAQUETARIA. El daño endotelial parece evidente, como lo demuestra la elevada concentración plasmática de proteínas liberadas desde las células endoteliales,(trombomodulina, factor von Willebrand y activador tisular del plasminógeno) durante el episodio agudo, con normalización durante la remisión20. También es posible detectar células endoteliales circulantes cuya concentración se correlaciona con el curso clínico de la enfermedad 21.
Moake 22 propone como mecanismo patogénico la presencia en plasma de factores agregantes de las plaquetas, que estarían constituidos por multímeros de Factor von Willebrand (FvW) de peso molecular inusualmente alto, los cuales pueden ser liberados a la circulación desde células endoteliales dañadas o estimuladas por autoanticuerpos, complejos inmunes o toxinas.
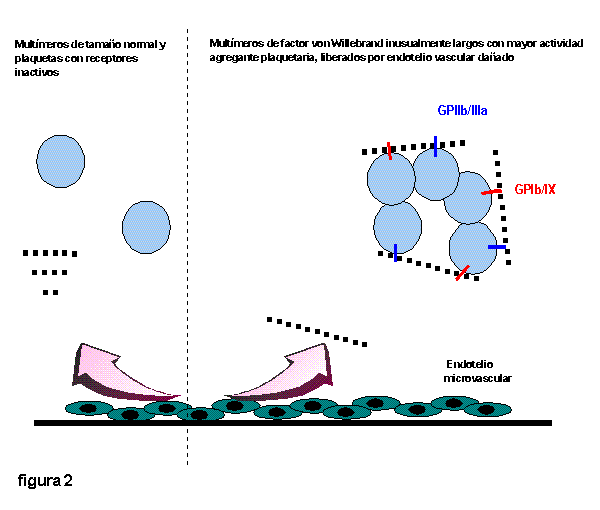
En condiciones normales, la célula endotelial sintetiza monómeros de FvW que se unen dando lugar a multímeros de elevado peso molecular. Estos multímeros son almacenados y posteriormente liberados desde la célula endotelial a la circulación. En el plasma son despolimerizados y convertidos en multímeros de menor tamaño por una proteína que podría estar ausente en los pacientes con PTT 23. Estos multímeros de FvW de peso molecular inusualmente alto poseen una mayor capacidad de agregación y adhesión plaquetaria (Figura 1) 22, 23. Se encontraron en pacientes portadores de PTT crónica en remisión y se piensa que no pueden detectarse durante la fase aguda de la enfermedad debido a su consumo en la agregación plaquetaria22, 23, 24(Figura 2).
Estos hallazgos sustentan la base fisiopatológica del tratamiento con infusión de plasma (el cual proporcionaría la actividad despolimerizante de las moléculas inusualmente grandes de FvW) y un soporte teórico para el uso de plasma sobrenadante de crioprecipitados (el cual está deplecionado de FvW).
Por otra parte, numerosos estudios también implican en la fisiopatogenia de la PTT a diversas alteraciones del metabolismo de la prostaciclina (derivado del ácido araquidónico, sintetizado por la célula endotelial y con actividad antiagregante plaquetaria). El plasma de algunos pacientes con PTT es incapaz de estimular la síntesis de prostaciclina 28, la cual parece estar también ausente en biopsias vasculares de pacientes afectos 29. Esta deficiencia se correlaciona con el hallazgo de bajas concentraciones plasmáticas durante el episodio agudo de un metabolito estable de la prostaciclina (6-cetoprostaglandin F1a), con normalización durante la remisión, y que revierte transitoriamente con la plasmaféresis 30.
La inhibición de la síntesis de prostaglandina podría ser debida a la liberación de b -Tromboglobulina, (proteína contenida en los gránulos a de las plaquetas) secretada tras la activación plaquetaria 31; o bien debida a una excesiva degradación de la misma, por la ausencia de un factor estabilizante 32.
El estado de hiperagregabilidad plaquetaria, junto con el hallazgo de plaquetas como principal componente de los microtrombos y el beneficio de los antiagregantes plaquetarios en el tratamiento de PTT, parecen implicar a una alteración plaquetaria primaria en el origen del proceso 33. Sin embargo no se ha demostrado activación de las plaquetas circulantes ni alteración de dicha actividad in vitro por agonistas 34.
Aunque todos los factores mencionados pueden estar implicados en la patogénesis de la PTT, su especificidad es discutida, ya que se manifiestan también en otras enfermedades asociadas con daño endotelial. Los multímeros FvW inusualmente grandes pueden ser detectados en pacientes con esclerodermia, LES y en cultivos de células endoteliales de pacientes con ricketsiosis 35. Asimismo, niveles plasmáticos elevados de trombomodulina y endotelina se objetivan en pacientes con CID y en el fallo cardiaco congestivo 36.
En conclusión, la gran heterogeneidad en los mecanismos patogénicos involucrados concuerda con un concepto de PTT-SHU entendido como un síndrome de etiología muy diversa, con una clínica común, como consecuencia de un daño endotelial difuso y trombos plaquetarios diseminados.
El aislamiento del serotipo E. Coli O157:H7, productor de verotoxina, junto con la identificación de la toxina libre en muestras de heces en 24 de 40 niños afectos de SHU, puso de manifiesto la relación entre dicha infección y el SHU infantil. Posteriormente numerosos estudios epidemiológicos han detectado la verotoxina o cepas enteropáticas de E. Coli en un 30-80% de los pacientes 37, 38, 39.
Alrededor del 90% de los casos de SHU ocurren en la infancia, lo cual sugiere la adquisición de inmunidad frente a la verotoxina en niños mayores y adultos.
La toxina se denominó inicialmente verotoxina ya que su efecto citopático estaba limitado a un reducido número de líneas celulares, que incluía a las células vero (células renales del mono verde africano). La verotoxina E. Coli está estructuralmente relacionada con la toxina Shiga producida por Shigella dysenteriae 1 40. Ambas toxinas pueden unirse al mismo receptor glicolípido, el cual está presente en células del endotelio vascular humano 41 y podrían ser las causantes del daño endotelial que inicia el proceso. Asimismo estas toxinas homólogas pueden estimular también la secreción de interleukina-8 42 (IL-8), la cual es un potente activador y quimioatrayente de los neutrófilos. Los niveles de IL-8 están aumentados en niños con SHU y se correlacionan con cifras elevadas de neutrófilos circulantes, los cuales parecen correlacionarse a su vez con un peor pronóstico 43.