|
El progreso de la Medicina ha mejorado el pronóstico de
muchos de los enfermos ingresados en las Unidades de Cuidados Intensivos,
aunque por el contrario ha hecho que aparezcan nuevas patologías en estos
pacientes, especialmente aquellos que permanecen largo tiempo ingresados
en nuestra Unidades. Entre ellas están diversas complicaciones
neurológicas que afectan al sistema nervioso periférico. De todas ellas,
la polineuropatía del paciente crítico es la entidad mejor definida y de
la que conocemos con mayor precisión sus características clínicas y
diagnósticas. Las alteraciones de la placa neuromuscular y sobre todo la
miopatía, que frecuentemente coexiste con la polineuropatía del paciente
crítico, son las otras complicaciones del sistema nervioso periférico que
desarrollan los pacientes críticos.
|
1.
Polineuropatía del paciente crítico |
En 1984, Bolton y col.
[1]
comunicaron cinco pacientes críticos sépticos que habían desarrollado en
UCI una polineuropatía axonal aguda, acuñándose el término de
polineuropatía del paciente crítico (PPC). Es una degeneración axonal
primaria de fibras motoras y sensitivas que se acompaña de degeneración de
las fibras musculares como resultado de la denervación aguda que sufren.
Ocurre especialemente en pacientes críticos que desarrollan Síndrome de
Respuesta Inflamatoria Sistémica (SRIS) y sepsis grave con Síndrome de
Disfunción Multiorgánica (SDMO).
1.1
Incidencia
La incidencia es variable dependiendo fundamentalmente
del tipo de pacientes estudiados y del momento de realización del estudio
neurofisiológico (ENF). Witt y col. [2]
realizaron el primer estudio prospectivo en una cohorte de 43 pacientes
con sepsis y SDMO. Treinta de ellos (70%) fueron diagnosticados de PPC, si
bien solo la mitad presentaban manifestaciones clínicas.
Posteriores estudios realizados en pacientes críticos con sepsis grave de
más de 7-10 días de duración confirman que la incidencia de PPC en estos
enfermos se sitúa alrededor del 75% [3, 4]. Por otro lado, en el
postoperatorio de cirugía cardiaca, un tercio de los pacientes que
requieren ventilación mecánica por más de tres días desarrollan PPC [5].
1.2 Etiopatogenia
La etiología precisa de la PPC continúa sin ser
conocida. Desde el trabajo de Zochodne y col. realizado en 19
pacientes con PPC se conoce que no es debido a un déficit específico
vitamínico o nutricional [6] . No obstante, en los últimos años se ha avanzado de forma considerable en
el conocimiento de diversos factores asociados con el desarrollo de PPC.
Cuando se estudia un grupo heterogéneo de 98 pacientes
críticos, se comprueba que la presencia de SRIS y la mayor gravedad de los
enfermos son los factores independientemente relacionados con la aparición
de PPC [7] .
Sin embargo, al analizar un grupo con sepsis grave se
observa que no todos los pacientes presentan esta complicación. Por ello,
se ha intentado determinar qué factores están relacionados con su
aparición. Witt y col. [2] mostraron que la presencia de PPC se
asociaba a la presencia de hiperglucemia e hipoalbuminemia, aunque se ha considerado que estos parámetros serían
marcadores de gravedad más que verdaderos agentes etiológicos. No
obstante, otros dos estudios posteriores han corroborado la posible
asociación entre trastornos metabólicos y el desarrollo de PPC.
En una cohorte de 73 pacientes críticos con sepsis
grave y ventilación mecánica durante más de 10 días, hemos evaluado los
factores de riesgo asociados a la PPC. Mediante un análisis multivariante,
los factores independientes para el desarrollo de PPC fueron la
hiperosmolaridad, el empleo de nutrición parenteral, el uso de relajantes
musculares y el fallo neurológico (definido como puntuación de Glasgow para
el coma inferior a 10 puntos), mientras que el empleo de técnicas de
depuración extrarrenal era un factor protector [4].
Posteriormente, un estudio prospectivo, aleatorizado y
controlado por placebo diseñado para evaluar la acción del tratamiento
insulínico intensivo con objeto de mantener una glucemia entre 80-110 mg/dL
en paciente críticos quirúrgicos demostró una reducción significativa en
la aparición de PPC en el grupo de tratamiento frente al grupo de
tratamiento convencional (51,9% frente a 28,7; p<0,001) [8] .
En un análisis multivariante se hallaron como factores independientes
asociados con el desarrollo de PPC: tratamiento convencional con insulina,
tratamiento con fármacos vasopresores por más de tres días, bacteriemia y
empleo de terapia de reemplazo renal. Esta disminución en la incidencia
de PPC se observó incluso cuando se comparó el grupo de pacientes con
glucemia entre 80-110 mg/dL con aquellos en que las cifras se mantuvieron
entre 100-150 mg/dL y esta reducción estuvo en relación con las cifras de
glucemia y no con la dosis de insulina aportada [9].
Por otro lado, un agente tóxico de bajo peso molecular
no bien identificado ha sido detectado en sangre de los pacientes que
presentan PPC y se sugiere que participa en el daño neuronal que estos
enfermos sufren [10] .
En un estudio multicéntrico reciente que incluyó 95
pacientes críticos tras al menos 7 días de ventilación mecánica y que
estaban conscientes para iniciar la desconexión del respirador, los
factores independientes para desarrollar una neuropatía axonal aguda
fueron el sexo femenino, el número de días con disfunción de dos o más
órganos, la duración previa de la ventilación mecánica y la administración
de corticoides [11] .
1.3 Anatomía patológica
En la histología se observan cambios compatibles con
una axonopatía sin afectación de la mielina ni signos inflamatorios [6,
12] .
No obstante, la biopsia nerviosa no es en absoluto necesaria para el
diagnóstico de la PPC.
Hay que reseñar un muy interesante estudio realizado en
24 pacientes por Latronico y col. [13] ,
los cuales mostraron que en algunos pacientes con diagnóstico
neurofisiológico de PPC la biopsia nerviosa no mostró alteraciones
morfológicas. Este hallazgo sugiere que los trastornos funcionales del
nervio pueden preceder a los cambios estructurales. Sin embargo, la
miopatía apareció en 23 de los 24 pacientes con diversos cambios
histológicos inespecíficos como ha sido puesto de manifiesto en otros
estudios que han realizado biopsia muscular en pacientes críticos [11,
14].
1.4 Diagnóstico
La PPC se manifiesta clínicamente cuando el paciente
mejora y se inicia la desconexión del respirador. Los reflejos osteo-tendinosos
suelen estar abolidos si bien podemos hallarlos reducidos o incluso
normales.
Los niveles séricos de creatinfosfoquinasa (CPK) son
normales o ligeramente elevados. El LCR no presenta alteraciones
patológicas. El diagnóstico diferencial debemos establecerlo con otros
procesos que cursan con debilidad muscular y que se presentan al ingreso
en UCI pero que pueden pasar desapecibidos por la gravedad inicial del
proceso que motiva ingreso en la Unidad (Tabla I) y los adquiridos en UCI
que cursan igualmente con debilidad generalizada (Tabla II).
Tabla I. Patología neuromuscular previa
al ingreso en UCI
-
Patología medular:
Traumatismo
Compresión medular (neoplasia, hematoma...)
Mielopatía isquémica
Mielitis transversa
Esclerosis lateral amiotrófica
-
Patología del sistema nervioso
periférico:
Síndrome de Guillain-Barré
Vasculitis
Polineuropatía diabética
Porfiria
Polineuropatía urémica
VIH
Neuropatía nutricional (tiamina, vitamina E...
Polineuropatía por fármacos: metronidazol, amiodarona, hidralacina,
nitrofurantoina, fenitoina, dapsona, cisplatino, vincristina, linezolid
Intoxicación por talio
-
Patología de la unión
neuromuscular:
Miastenia gravis
Intoxicación por organofosforados
Botulismo
parálisis por picadura de garrapata
Síndrome de Lambert-Eaton
Toxicidad por aminoglucósidos
Toxicidad por colistina
Hipermagnesemia
-
Patología muscular:
Distrofia muscular
Polimiositis
Miopatía mitocondrial
Tabla II. Debilidad adquirida en UCI
-
Trastornos del sistema nervioso
periférico:
Polineuropatía del paciente crítico
Síndrome de Guillain-Barré
Neuropatía nutricional
Polineuropatía por fármacos
Neuropatía motora por bloqueantes neuromusculares
-
Trastornos de la unión
neuromuscular:
Toxicidad por aminoglucósidos
Toxicidad por colistina
Parálisis persistente por bloqueantes neuromusculares
Hipermagnesemia
-
Patología muscular:
Mioptía del paciente crítico
Miopatía de filamento grueso
Miopatía necrotizante
Para el diagnóstico de la PPC es imprescindible un ENF
que comprenda la realización de un electroneurograma y un electromiograma
[15] .
En el ENF se observa degeneración axonal primaria de fibras motoras y
sensitivas con atrofia por denervación de los músculos esqueléticos, si
bien algunos autores han encontrado una afectacion predominante de las
fibras motoras [16]. Los datos
propios de una degeneración axonal son la conservación de la velocidad de
conducción, latencia distal normal y la caída del potencial de respuesta
motora, que puede llegar a desaparecer en los casos graves. Es muy
importante para el diagnóstico la búsqueda de denervación en los músculos
(Figura 1), como expresión de la agudeza del proceso que se manifiesta por
aparición en el EMG de potenciales de fibrilación y ondas positivas
[1.
Figura 1. Registro típico de denervación en una
axonopatía aguda: potenciales de fibrilación y ondas positivas
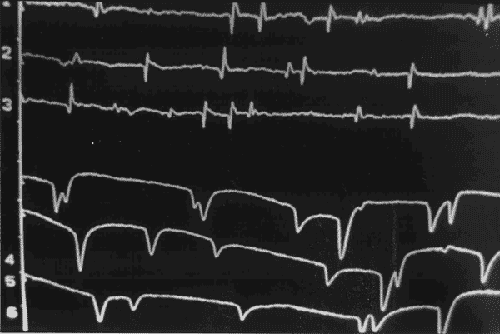
Debemos igualmente realizar una estimulación repetitiva
a 3 y 20 Hz para explorar la unión neuro-muscular y descartar la
persistencia de un bloqueo de conducción a dicho nivel, bien por fármacos
o bien por trastornos metabólicos (ver Tabla II). En la PPC no se detectan
bloqueos de conducción. Estos cambios pueden aparecer de forma precoz, ya
que se ha comprobado que una reducción patológica del potencial de
respuesta motora se detecta a los 2-5 días tras el ingreso en UCI [18] .
1. 5
Tratamiento
Actualmente no existe tratamiento efectivo para la
PPC. Como excepción, un estudio retrospectivo encontró que los
pacientes que habían recibido tratamiento con inmunoglobulinas para la
sepsis presentaban una incidencia más baja de PPC que
aquéllos a los que no se les administró este tratamiento [19] .
Por otro lado, se ha comunicado que el empleo de inmunoglobulinas a dosis
similares que en el síndrome de Guillain-Barré no modifica la evolución en
aquellos sujetos que han desarrollado ya una PPC [20].
El tratamiento actual pasa por la identificación del
problema y la instauración de fisioterapia precoz. Por otro lado, la
identificación de los factores de riesgo nos debe llevar a evitar el uso
de bloqueantes neuromusculares y a un estrecho control metabólico de los
pacientes sépticos, evitando especialmente la hiperglucemia y la
hiperosmolaridad.
1. 6
Pronóstico
En los casos de polineuropatía leve la recuperación es
favorable en semanas. En los casos de afectación grave, el pronóstico
funcional no es bueno, persistiendo a los dos años una importante
limitación de la movilidad y una calidad de vida muy deteriorada. La
coexistencia de una neuropatía axonal con elentecimiento de la velocidad
de conducción se asocia a una peor recuperación [21] .
Una prolongada estancia en UCI, mayor duración de la sepsis y la pérdida
de peso son los tres parámetros que se asocian a una peor recuperación
según un estudio reciente que siguió la evolución durante dos años de 19
pacientes con PPC [22].
Recientemente, dos estudios han confirmado la
persistencia de la afectación neurológica periférica durante años.
Herridge y col. [23]
han estudiado 109 supervivientes de un síndrome de distrés respiratorio
agudo a los 12 meses del alta de UCI. Estos pacientes presentaron SDMO, y a
los 12 meses existía una grave debilidad muscular, lo que se puso de
manifiesto por el test de la distancia caminada en 6 minutos.
Por su parte, Fletcher y col. [24]
han evaluado 22 pacientes que habían permanecido más de 28 días ingresados
en UCI. Dicho estudio se realizó una mediana de 42,5 meses tras el alta y
los pacientes habían permanecido en ventilación mecánica por una mediana
de 37 días. Los hallazgos más destacados de este trabajo fueron que el 100%
de los enfermos continuaban con clínica de debilidad importante y que en
el estudio neurofisiológico existían datos indicativos de neuropatía axonal (PPC) en 21 de los 22 enfermos, siendo
infrecuentes los datos de miopatía.
|
2. Miopatía del paciente crítico |
Los primeros casos de miopatía en pacientes críticos
fueron descritos en la década de los setenta tras el empleo de altas dosis de
corticoides y bloqueantes neuromusculares, acuñándose el término de miopatía
tetrapléjica aguda [25] .
No es hasta la década de los 90 cuando se describen pacientes críticos,
generalmente con sepsis y SDMO, que desarrollan una miopatía aguda pero
sin estar en relación con el empleo de estos fármacos.
2 .1
Clasificación
Se han realizado diversas clasificaciones
histológicas de la miopatía aguda en el paciente crítico. Recientemente,
Hund propuso la clasificación de miopatía del paciente crítico, miopatía
de filamento grueso y miopatía necrotizante, basada en los cambios
histológicos presentes
[26].
2.2 Fisiopatología
Los mecanismos íntimos no son bien conocidos, habiéndose
propuesto muy diversos factores que pueden contribuir al desarrollo de miopatía en el paciente crítico. Por un lado, tendríamos la repercusión de
la sepsis sobre el músculo y por otro lado el papel de los
corticosteroides y bloqueantes neuromusculares no despolarizantes (BNM-ND), que debido al aumento de la permeabilidad vascular
que existe en la sepsis fácilmente pueden acceder y dañar al músculo.
En el músculo, existen cuatro sistemas proteolíticos:
complejo ubiquitina-proteasoma, proteasas lisosomales, proteasas calcio
dependientes (calpaína) y proteasas no lisosomales que no dependen de
calcio o ATP. Diversos mediadores proinflamatorios implicados en la sepsis
(TNF e IL-1 principalmente) inducen proteolisis. Estas citocinas activan
la ubiquitina-proteasoma que es la principal vía intracelular de
degradación proteica. En la sepsis se ha comprobado que se produce una
activación de esta vía, como ha sido demostrado en humanos [27] .
Por el contrario, en cinco pacientes con miopatía del paciente crítico
(tres había recibido esteroides) no se demostró activación de la
ubiquitina, siendo la proteolisis mediada por la activación de la calpaína
y no por activación de las otras vías [28].
Recientemente, se ha demostrado en un modelo animal de
sepsis que la debilidad muscular que presentaban estas ratas se asociaba a
la presencia de anticuerpos contra el receptor de la acetilcolina, al igual
que ocurre en la miastenia gravis
[29].
Además, aunque la ausencia de infiltrado inflamatorio
es casi constante en la microscopía, se encontró la presencia de
marcadores inmunohistoquímicos de inflamación en 3 de 5 pacientes con el
diagnóstico de miopatía del paciente crítico, sugiriendo que en la
patogenia de esta entidad puede participar un mecanismo
inflamatorio [30].
2.3 Anatomía patológica
En la miopatía del paciente crítico la histología
revela atrofia predominantemente de las fibras musculares del tipo II,
miofibrillas sin cambios inflamatorios, fibrosis aislada y necrosis
muscular ausente o limitada a fibras aisladas. Ocurre
principalmente en pacientes con sepsis grave y SDMO. Frecuentemente estos
cambios se asocian a la PPC [13.
La miopatía de filamento grueso afecta a pacientes que
han recibido altas dosis de esteroides, usualmente en combinación con BNM-ND.
En la miopatía necrotizante existe mionecrosis extensa y fagocitosis de
las fibras musculares, afectando predominantemente a pacientes que han
recibido corticosteroides y/o BNM-ND. No obstante, esta miopatía también
puede afectar a pacientes con sepsis grave y SDMO [31].
2.4 Incidencia
La incidencia de miopatía del paciente crítico no es
bien conocida. La principal razón es que la miopatía es difícil de
detectar, ya que los estudios neurofisiológicos no son sensitivos ni
específicos, y se requiere un procedimiento invasivo como es la biopsia
muscular. Varios estudios prospectivos han apuntado que la mayoría de los
pacientes con SDMO y estancia prolongada en UCI tienen cambios patológicos
en la biopsia muscular. Existen únicamente 5 estudios prospectivos que
describen una serie de biopsias en pacientes críticos sin enfermedad
muscular previa conocida [13,14 ,
31-33]. La incidencia de necrosis muscular varía considerablemente
(9-55%), así como la de atrofia (39-96%), probablemente por los diferentes
criterios de inclusión entre los distintos estudios.
Así, en una serie de 31 pacientes críticos con biopsia
muscular, 15 presentaban miopatía necrotizante. La mayoría de estos
pacientes presentaban criterios clínicos de sepsis pero no se conoce
cuántos de ellos habían recibido corticoides y/o BNM-ND [31.
Nosotros hemos observado dos pacientes sépticos con sepsis grave y SDMO
que desarrollaron miopatía necrotizante grave confirmada por histología y
que no habían recibido corticosteroides ni BNM-ND [34] .
Lacomis y col. estudiaron 92 pacientes
críticos con electromiografía [32. La
causa más frecuente de debilidad en UCI fue la miopatía, diagnosticada en
39 de ellos. Hay que reseñar que la mayoría de estos pacientes eran
trasplantados de órganos sólidos que habían recibido elevadas cantidades de
esteroides, no conociéndose cuántos habían desarrollado sepsis. En 14 de
estos pacientes se realizó biopsia muscular, que reveló miopatía con
pérdida de miosina (filamento grueso) en el 64% de ellos.
2.5 Manifestaciones clínicas
El cuadro clínico de una miopatía es indistinguible de
otras causas de enfermedad neuromuscular adquirida en UCI. Los
pacientes críticos con miopatía presentan debilidad simétrica de miembros
y músculos respiratorios. Los reflejos profundos suelen estar reducidos o
ausentes.
2.6 Diagnóstico
La CPK sérica está dentro del rango de la normalidad o
ligeramente elevada, excepto en los casos de miopatía necrotizante, que
generalmente tienen una marcada elevación de la misma.
El diagnóstico se basa en el ENF y en la biopsia
muscular. El EMG con aguja registra el patrón de actividad en el músculo
tanto en reposo como en actividad. Los dos signos que claramente pueden
diferenciar entre neuropatía y miopatía son el análisis del potencial de
acción de unidad motora (PAUM) y el reclutamiento de fibras generado por el
esfuerzo voluntario. Para ello, los pacientes deben cooperar y realizar
una contracción voluntaria. Los PAUM son normalmente bifásicos y con una
duración de entre 5 y 15 ms. Tras la denervación, como ocurre en la PPC,
los PAUM aumentan de amplitud y duración, con reclutamiento aumentado. Estos
cambios pueden persistir años. En las miopatías, por el contrario, los PAUM
están disminuidos en amplitud y duración y son polifásicos, existiendo
reclutamiento precoz [15].
Obviamente, dado que para la obtención de PAUM se
requiere la cooperación del paciente puede existir una infraestimación de
la frecuencia de la miopatía en pacientes críticos. Por otro lado, el EMG
no ayuda al diagnóstico etiológico de la miopatía, y no diferencia entre
sus diversas causas.
La biopsia muscular es el método diagnóstico de
elección para la detección de anomalías estructurales, pero es un
método invasivo y puede no ser reproducido con facilidad. Actualmente se
debería realizar la biopsia muscular en aquellos pacientes con debilidad
muscular, especialmente si impide la desconexión de la ventilación
mecánica, sin un diagnóstico neurofisiológico de PPC ni bloqueo de
conducción en el ENF.
2.7 Tratamiento
No existe tratamiento específico. La mayoría de los
autores están de acuerdo en que debería evitarse el empleo de dosis altas
de BNM-ND y que cuando fueran necesarios, deberían administrarse en bolos
mejor que en perfusión continúa.
-
Bolton CF, Gilbert JJ, Hahn
AF, Sibbald WJ. Polyneuropathy in critically ill patient. J Neurol
Neurosurg Psych 1984; 47: 1223-1231.
-
Witt NJ, Zochodne DW,
Bolton CF, Maison FG, Wells G, Young B, et al. Peripheral nerve function
in sepsis and multiple organ failure. Chest 1991; 99: 176-184.
-
Berek K, Margreiter J,
Willeit J, Berek K, Schmutzhard E, Mutz NJ. Polyneuropathies in
critically ill patients: a prospective evaluation. Intensive Care Med
1996; 22: 849-855.
-
Garnacho-Montero
J, Madrazo-Osuna J, García Garmendia JL, Ortiz Leyba C, Jiménez-Jiménez
FJ, Barrero-Almodóvar AE, Garnacho-Montero MC, Moyano del Estad MR.
Ctritical illness polyneuropathy: Risk factors and clinical
consequences. A cohort study in septic patients. Intensive Care Med
2001; 27: 1288-1296.
-
Thiele RI, Jakob H, Hund E, et
al. Sepsis and
catecholamine support are the major risk factors for crtical illness
polyneuroapthy after open heart surgery. Thorac Cardiovasc Surg 2000;
48: 145-150.
-
Zochodne DW, Bolton CF,
Wells GA, et al. Critical illness polyneuropathy. A complication of
sepsis and multiple organ failure.
Brain 1987; 110: 819-842.
-
De Letter MA, Schmitz PI,
Visser LH, Verheul FAM, Schellens RLLA, Op de Coul DAW, van der Merché
FGM. Risk factors
for the development of polyneuropathy and myopathy in critically ill
patients. Crit Care Med 2001; 29:
2281-2286.
-
Van den Berghe G, Wouters
P, Weekers F, et al. Intensive insulin therapy in critically ill
patients. N Engl J Med 2001; 345: 1359-1367.
-
Van der Berghe G, Wouters
PJ, Bouillon R, et al. Outcome benefit of intensive insulin therapy in
the critically ill: Insulin dose versus
glycemic control. Crit Care Med 2003; 31: 359-366.
-
Druschky A, Herkert M,
Radespiel-Tröger M, et al. Critical illness polyneuropathy: clinical
findings and cell culture assay of neurotoxicity assessed by a
prospective study.
Intensive Care Med 2001; 27: 686-693.
-
De Jonghe B, Sharshar T,
Lefaucheur JP, et al.
Paresis acquired in the
intensive care unit. A prospective multicenter study. JAMA 2002; 288;
2859-2867.
-
Hund EF, Fogel W, Krieger
D, DeGeorgia M, Hacke W. Critical illness polyneuropathy: Clinical
findings and outcomes of a frequent cause of neuromuscular weaning
failure. Crit Care Med 1996; 24:
1328-1333.
-
Latronico N, Fenzi F, Recupero D, et al.
Critical illness
myopathy and neuropathy. Lancet 1996; 347: 1579-1582.
-
Coakley JH, Nagendran K,
Yarwood GD, et al. Patterns of neurophysiological abnormalities in
proloned critical illness. Intensive Care Med 1998; 24: 801-807.
-
Bolton CF.
Electrophysiologic studies of critically ill patients. Muscle Nerve
1987; 10: 129-135.
-
Hund E, Genzwurker H,
Bohrer H, et al. Predominant involvement of the motor fibres in patients
with critical illness polyneuropathy. Br J Anaesthesia 1997; 78:
274-279.
-
Bolton CF. Sepsis and the
systemic inflammatory response syndrome: neuromuscular manifestations.
Crit Care Med 1996; 24: 1408-1416.
-
Tennilä A, Salmi T, Perrilä
V, Roine RO, Varpula T, Takkunen O. Early signs of critical illness
polyneuropathy in ICU patients with systemic inflammatory response
syndrome or sepsis. Intensive Care Med 2000; 26: 1360-1363.
-
Mohr M, Englisch L, Roth A,
Burchardi H, Zielmann S. Effects of early treatment with immunoglobulin
on critical illness polyneuropathy following multiple organ failure and
gram-negative sepsis. Intensive Care Med 1997; 23:
1144-1149.
-
Wijdicks EFM, Fulgham JR.
Failure of high dose intraveous immunoglobulins to alter the clinical
course of critical illness polyneuropathy. Muscle Nerve 1994; 17:
1494-1495.
-
Leijten FSS, Harinck-De
Weerd JE, Poortvliet DCJ, De Weerd AW. The role of polyneuropathy in
motor convalescence after prolonged mechanical ventilation.
JAMA 1995; 274: 1221-1225.
-
De Seze M, Petit H, Wiart L,
et al. Critical
illness polyneuropathy. A 2-year folow-up study in 19 severe cases. Eur
Neurol 2000; 43: 61-69.
-
Herridge MS, Chenung AM,
Tansey CM, Matte-Martyn A, Díaz-Granados N, Al-Saidi F, et al. One-year
outcomes in survivors of acute respiratory distress syndrome. N Engl J
Med 2003; 348: 683-693.
-
Fletcher SN, Kennedy DD,
Ghosh IR, Misra VP, Kiff K, Coakley JH, et al. Persistent neuromuscular
and neurophysiologic abnormalities in long-term survivors of prolonged
critical illness. Crit Care Med 2003; 31: 1012-106.
-
MacFarlane IA, Rosenthal
FD. Severe myopathy after status asthmaticus. Lancet 1977; 2:
615.
-
Hund E. Myopathy in
critically ill patients. Crit Care Med 1999; 27: 2544-2547.
-
Tiao G, Hobler S, Wang JJ,
Meyer TA, Luchette FA, Fischer JE, et al.
Sepsis is associated with
increased mRNAs of ubiquitin-proteasome proteolytic pathway in human
skeletal muscle. J Clin Invest 1997; 99: 163-168.
-
Showalter CJ, Engel AG.
Acute quadriplegic myopathy: Analysis of myosin isoforms and evidence
for calpain-mediated proteolysis. Muscle Nerve 1997; 20: 316-322.
-
Tsukagoshi H, Morita T,
Takahashi K, Kunimoto F, Goto F. Cecal ligation and puncture peritonitis
model shows a decrease nicotinic acetylcholine receptor numbers in rat
muscle: Immunopathologic mechanisms? Anesthesiology 1999; 91: 448-460.
-
Bazzi P, Moggio M, Prelle
A, Sciacco M, Messina S, Barberini S, et al. Critically ill patients:
immunological evidence of inflammation in muscle biopsy. Clin Neuropath
1999; 18: 23-30.
-
Helliwell TR, Coakley JH,
Wagemakers AJM et al. Necrotizing myopathy in critical ill patients.
Journal of Pathology 1991; 164:
307-314.
-
Lacomis D, Petrella JT, Giuliani MJ.
Causes of
neuromuscular weakness in the intensive care unit: A study of ninety-two
patients. Muscle Nerve 1998; 21: 610-617.
-
Coakley JH, Nagendran K,
Honavar M, Hinds CJ. Preliminary observations on the neuromuscular
abnormalities in patients with organ failure and sepsis. Intensive Care
Med 1993; 19:
323-328.
-
Garnacho-Montero J, Jiménez-Jiménez FJ,
Barrero-Almodóvar AE, Amaya-Villar R, Luque R, García-Garmendia Jl, et
al. Necrotizing
myopathy in patients with severe sepsis. Intensive care Med 2000; 26:
S247.
José Garnacho Montero, Carlos Ortiz Leyba, Rosario
Amaya Villar
Hospital Virgen del Rocío, Sevilla
©REMI,
http://remi.uninet.edu. Diciembre 2004.
Palabras clave:
Sepsis, Sepsis grave, Enfermedad neuromuscular del enfermo crítico,
Polineuropatía del enfermo crítico, Miopatía del enfermo crítico.
Busque en REMI con Google:
Envía tu comentario para su
publicación |



